Adenine Base Editors (ABEs): A Complete Guide to Precision Genome Editing Mechanisms and Applications
This comprehensive article provides researchers, scientists, and drug development professionals with an in-depth analysis of adenine base editors (ABEs).
Adenine Base Editors (ABEs): A Complete Guide to Precision Genome Editing Mechanisms and Applications
Abstract
This comprehensive article provides researchers, scientists, and drug development professionals with an in-depth analysis of adenine base editors (ABEs). We explore the foundational molecular architecture of ABEs, detailing their fusion of catalytically impaired Cas9 with engineered deaminase enzymes. The article systematically covers the core methodology for deploying ABEs in various cellular models and key therapeutic applications. We address common experimental challenges, including off-target effects and delivery optimization, and provide troubleshooting strategies. Finally, we present a comparative analysis of ABE variants and current validation frameworks. This resource synthesizes the latest research to guide the effective design and implementation of ABE technology in biomedical research and therapeutic development.
What Are Adenine Base Editors? Unpacking the Molecular Machinery of A•T to G•C Conversion
This whitepaper details the core principles of precision genome editing, tracing the evolution from foundational CRISPR-Cas9 systems to the highly specific mechanisms of Adenine Base Editors (ABEs). Framed within the research thesis "How do adenine base editors (ABEs) work?", this guide provides a technical dissection of ABE architecture, kinetics, and experimental application for researchers and drug development professionals.
From CRISPR-Cas9 to Base Editing: A Paradigm Shift
CRISPR-Cas9 revolutionized genetics by enabling targeted DNA double-strand breaks (DSBs), which are subsequently repaired by cellular machinery. However, this reliance on endogenous repair pathways—predominantly Non-Homologous End Joining (NHEJ) or Homology-Directed Repair (HDR)—introduces inefficiencies and unpredictable indels. Base editors represent a paradigm shift, directly converting one target DNA base pair to another without creating a DSB, thereby minimizing unintended mutagenesis.
Core Architectural Evolution:
- CRISPR-Cas9: Single guide RNA (sgRNA) + Cas9 endonuclease → DSB → Cellular Repair.
- Cytosine Base Editor (CBE): sgRNA + nickase Cas9 (nCas9/D10A) + Cytidine Deaminase → C•G to T•A conversion.
- Adenine Base Editor (ABE): sgRNA + nickase Cas9 (nCas9/D10A) + Engineered Adenine Deaminase → A•T to G•C conversion.
The Core Mechanism of Adenine Base Editors (ABEs)
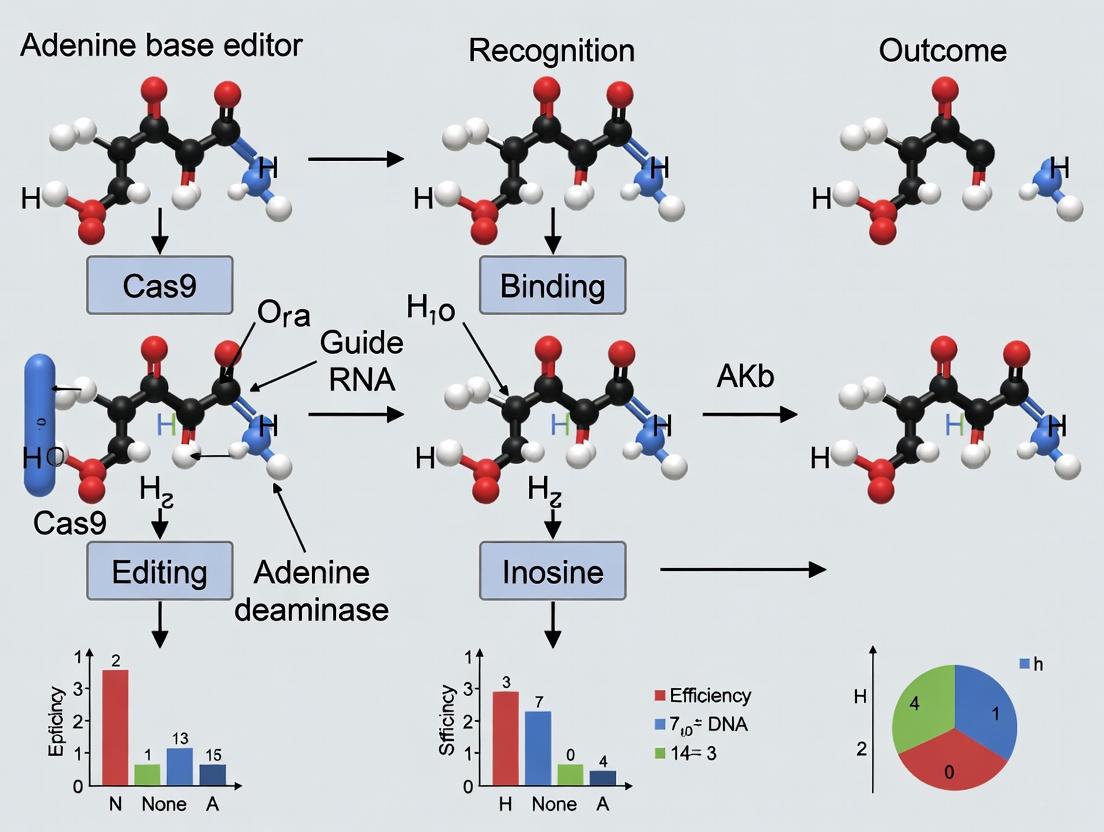
ABEs are fusion proteins that combine a catalytically impaired Cas9 (nCas9, which nicks the non-edited strand) with an engineered adenine deaminase enzyme (e.g., TadA-8e). The system is guided to a specific genomic locus by an sgRNA. Within the sgRNA-defined protospacer, the deaminase catalyzes the hydrolytic deamination of adenine (A) to inosine (I) in the single-stranded DNA bubble formed by Cas9. Inosine is read as guanine (G) by cellular polymerases. The nick in the complementary strand triggers cellular repair, which replaces the thymine (T) with a cytosine (C), completing the A•T to G•C base pair conversion.
Quantitative Performance Metrics of Current ABE Variants
Data sourced from recent literature (2023-2024).
Diagram Title: ABE Core Reaction Pathway
Table 1: Performance Characteristics of Advanced ABE Variants
| Editor Name | Deaminase Variant | Target Window (Proto-spacer Position) | Typical Editing Efficiency (%) | Product Purity (% G•C)* | Key Reported Indel Rate (%) |
|---|---|---|---|---|---|
| ABE8e | TadA-8e | 4-8 (SpCas9) | 30-60 | >99.9 | <0.1 |
| ABE8.8 | TadA-8.8 (V106W) | 4-8 (SpCas9) | 35-70 | >99.9 | <0.1 |
| ABE9e | TadA-9e | 4-10 (SpCas9) | 20-50 | >99.5 | <0.5 |
| ABEmax | TadA-8e | 4-7 (SpCas9) | 40-80 | >99.9 | <0.1 |
| miniABEmax | TadA-8e | 4-7 (SaCas9-KKH) | 15-40 | >99.5 | <0.5 |
*Product Purity: Ratio of desired base conversion to indels/other byproducts.
Experimental Protocol: In Vitro Validation of ABE Activity
Objective: To quantify the on-target editing efficiency and product distribution of an ABE construct at a defined genomic locus in cultured mammalian cells.
Methodology:
1. Design & Cloning:
- Design sgRNA (20-nt spacer) targeting the locus of interest. Ensure an adenine (A) is present in the correct strand within positions 4-10 of the protospacer (for SpCas9-based ABEs).
- Clone sgRNA sequence into an appropriate expression plasmid (e.g., U6-driven).
- Obtain plasmid expressing the ABE protein (e.g., ABEmax) under a constitutive or inducible promoter.
2. Cell Transfection:
- Culture HEK293T or other relevant cell line.
- Co-transfect cells with the ABE expression plasmid and sgRNA plasmid using a polyethylenimine (PEI) or lipid-based method. Include a transfection control (e.g., GFP plasmid).
- Harvest cells 72-96 hours post-transfection.
3. Genomic Analysis:
- Genomic DNA Extraction: Use a silica-column-based kit.
- PCR Amplification: Amplify the target region (∼300-500 bp) using high-fidelity polymerase.
- Sequencing & Analysis:
- Sanger Sequencing: Clean PCR products and sequence. Analyze chromatograms for dual peaks using decomposition software (e.g., EditR, BEAT).
- Next-Generation Sequencing (NGS): Perform a two-step PCR to add Illumina adapters and barcodes. Pool libraries and sequence on a MiSeq. Analyze reads using pipelines like CRISPResso2 or BEAT for precise quantification of base substitutions and indels.
Critical Controls: Include a "sgRNA-only" transfection control to assess background noise.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for ABE Research
| Reagent / Material | Function / Description | Example Vendor/Product |
|---|---|---|
| ABE Expression Plasmid | Delivers the ABE fusion protein (nCas9-TadA). | Addgene #112095 (pCMV-ABEmax) |
| sgRNA Cloning Vector | Backbone for expressing sgRNA under RNA Pol III promoter. | Addgene #41824 (pU6-sgRNA vector) |
| High-Efficiency Transfection Reagent | For delivering plasmids into mammalian cells. | Lipofectamine 3000 (Thermo), PEIpro (Polyplus) |
| Genomic DNA Extraction Kit | Rapid, pure gDNA isolation from cultured cells. | DNeasy Blood & Tissue Kit (Qiagen) |
| High-Fidelity PCR Polymerase | Accurate amplification of target genomic loci. | Q5 High-Fidelity DNA Polymerase (NEB) |
| NGS Library Prep Kit | For preparing amplicon sequencing libraries. | KAPA HiFi HotStart ReadyMix with unique dual indexing. |
| BE Analysis Software | Computational tool to quantify base editing from NGS data. | CRISPResso2, BEAT (Base Editing Analysis Tool) |
Advanced Considerations & Current Frontiers
Table 3: Addressing ABE Limitations - Technical Strategies
| Challenge | Underlying Cause | Emerging Solutions |
|---|---|---|
| Off-Target Editing | Deaminase activity on ssDNA at similar but non-identical sequences. | Use of high-fidelity Cas9 variants; Engineering deaminase mutants with narrowed activity windows. |
| Restricted Targeting Scope | Requires a PAM (NGG for SpCas9) and an A within the activity window. | Development of ABEs using Cas9 orthologs with alternative PAMs (e.g., SaCas9, Nme2Cas9). |
| Bystander Editing | Deamination of multiple A's within the activity window. | Optimization of linker length; Directed evolution of TadA variants with altered activity windows. |
| Delivery Constraints | Large size of SpCas9-based editors limits AAV packaging. | Development of compact ABEs using smaller Cas proteins (e.g., miniABEmax). |
Diagram Title: ABE Experimental Workflow
Adenine Base Editors are a direct technological descendant of CRISPR-Cas9, engineered to execute precise, efficient, and predictable A•T to G•C conversions. Their core principle hinges on the fusion of a programmable, nickase DNA-binding protein with an evolved adenine deaminase. While challenges in off-target editing, delivery, and targeting scope persist, ongoing protein engineering and mechanistic research continue to refine the platform. ABEs now serve as indispensable tools for functional genomics, disease modeling, and are progressing toward clinical applications for correcting pathogenic point mutations.
Adenine Base Editors (ABEs) represent a transformative class of precision genome editing tools that enable the direct, irreversible conversion of an A•T base pair to a G•C base pair without inducing double-strand DNA breaks (DSBs). This in-depth guide deconstructs the core components of an ABE—the Cas9 nickase, the adenine deaminase, and the critical linker architecture—framed within the central research thesis: How do adenine base editors (ABEs) work? Understanding this molecular machinery is crucial for researchers and drug development professionals aiming to develop therapeutic corrections for pathogenic point mutations, which constitute a majority of known human genetic disorders.
Core Component Architecture
Cas9 Nickase (nCas9)
The Cas9 nickase serves as the programmable DNA-targeting module. It is typically derived from Streptococcus pyogenes Cas9 (SpCas9) but engineered with a single-point mutation (D10A) in its RuvC nuclease domain. This mutation abolishes its ability to cleave the target DNA strand while retaining the ability to cleave (nick) the non-target, or "glycerol," strand. This targeted nick serves to bias cellular DNA repair pathways toward using the edited strand as a template, thereby increasing the efficiency of base editing and reducing the generation of indels.
Adenine Deaminase
The catalytic engine of the ABE is an evolved adenine deaminase. The first-generation ABEs utilized the E. coli TadA (tRNA-specific adenosine deaminase), which naturally acts on single-stranded RNA. Through multiple rounds of directed evolution, TadA was engineered to deaminate adenosine in single-stranded DNA. Modern ABEs (e.g., ABE8e) often use a heterodimer of wild-type TadA (wtTadA) and evolved TadA (eTadA*) to enhance stability and activity. The deaminase catalyzes the hydrolytic deamination of adenosine (A) to inosine (I), which is read as guanosine (G) by DNA polymerases during replication or repair.
Linker Architecture
The linker is a critical, often undervalued, structural component that connects the deaminase to the nCas9. It determines the spatial positioning and flexibility of the deaminase relative to the displaced single-stranded DNA (ssDNA) bubble created by Cas9 binding. The length, composition (e.g., Gly-Ser repeats), and rigidity of the linker directly influence the editing window—the span of nucleotides within the protospacer where A-to-G conversion can occur. Optimized linkers are essential for balancing editing efficiency, purity, and specificity.
Table 1: Evolution and Performance Metrics of Key ABE Variants
| ABE Variant | Deaminase Composition | Common Linker Length | Primary Editing Window (nt from PAM) | Typical Efficiency (in vitro) | Key Reference (Year) |
|---|---|---|---|---|---|
| ABE7.10 | TadA*-TadA (heterodimer) | 16-aa (XTEN) | Positions 4-8 | ~50% | Gaudelli et al., Nature (2017) |
| ABE8e | TadA*-TadA (heterodimer) | 16-aa (XTEN) | Positions 4-8 | Up to ~95% | Richter et al., Nature Biotechnology (2020) |
| ABE8s | TadA*-TadA (heterodimer) | 12-aa (Shorter) | Positions 4-7 | High, with reduced off-target | Gaudelli et al., Nature Biotechnology (2020) |
| ABE9 | TadA*-TadA (heterodimer) | Engineered Rigid | Position ~5 | High, >99% product purity | Chen et al., Nature (2023) |
Table 2: Impact of Linker Properties on Editing Outcomes
| Linker Type | Length (Amino Acids) | Flexibility/Rigidity | Effect on Editing Window Width | Effect on Off-Target Editing (DNA) | Primary Use Case |
|---|---|---|---|---|---|
| XTEN | 16-84 | Highly Flexible | Broadens (~5-9 nt) | Can increase | Standard ABE7.10/8e |
| (GGS)n | Variable (e.g., n=9) | Flexible | Tunable | Moderate | Custom tuning |
| Rigid α-helical | Fixed (e.g., 12-aa) | Rigid | Narrows and shifts | Often decreases | High-fidelity ABE9, targeting specific positions |
Detailed Experimental Protocol: Evaluating ABE Editing Efficiency and Specificity
Protocol: HEK293T Cell Transfection and NGS Analysis for ABE Characterization
Objective: To quantitatively assess the on-target editing efficiency and byproduct profile of a novel ABE variant at a defined genomic locus.
Materials: See "The Scientist's Toolkit" below.
Methodology:
- Plasmid Design: Clone the gene for the novel ABE variant (nCas9-D10A, linker, deaminase) into a mammalian expression plasmid (e.g., pCMV). Design and clone a single-guide RNA (sgRNA) expression plasmid targeting a standard locus (e.g., HEK site 3).
- Cell Culture: Maintain HEK293T cells in DMEM + 10% FBS at 37°C, 5% CO2.
- Transfection: Seed cells in a 24-well plate. At ~70% confluence, co-transfect 500 ng of ABE plasmid and 250 ng of sgRNA plasmid using 2 µL of polyethylenimine (PEI) reagent per well.
- Genomic DNA Harvest: 72 hours post-transfection, aspirate medium, lyse cells directly in the well with 200 µL of Direct Lysis Buffer (50 mM KCl, 10 mM Tris-HCl pH 8.3, 0.1% Triton X-100, 60 µg/mL Proteinase K). Incubate at 56°C for 1 hour, then 95°C for 10 minutes to inactivate Proteinase K.
- PCR Amplification: Use 2 µL of lysate as template in a 50 µL PCR reaction with high-fidelity polymerase to amplify the target genomic region (~300-400 bp surrounding the edit site). Include barcoded sequencing adapters in the primers.
- Next-Generation Sequencing (NGS) Library Prep: Purify PCR products, normalize concentrations, and pool samples. Perform a secondary limited-cycle PCR to add full Illumina flow cell adapters and indices.
- Data Analysis: Sequence on a MiSeq system. Process reads through a standard pipeline (e.g., CRISPResso2) to align sequences and quantify the percentage of A-to-G conversions at each position within the protospacer, as well as insertion/deletion (indel) frequencies.
Mandatory Visualizations
Diagram 1: ABE Complex Mechanism (100 chars)
Diagram 2: Linker Design Impacts Editing Window (100 chars)
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for ABE Research
| Item | Function/Description | Example Vendor/Cat. No. (Illustrative) |
|---|---|---|
| ABE Expression Plasmids | Mammalian codon-optimized vectors for nCas9, linker, and deaminase (e.g., ABE8e). Essential for delivery of editor. | Addgene (#138489 for ABE8e) |
| sgRNA Cloning Vector | Backbone for expressing single-guide RNA under a U6 promoter. | Addgene (#41824) |
| HEK293T Cells | Robust, easily transfected human cell line standard for initial editor benchmarking. | ATCC (CRL-3216) |
| Polyethylenimine (PEI) | High-efficiency, low-cost cationic polymer transfection reagent for plasmid DNA. | Polysciences (23966-1) |
| Direct Lysis Buffer | Allows rapid cell lysis and gDNA extraction without column purification for PCR. | In-house formulation (see protocol). |
| High-Fidelity PCR Polymerase | For accurate amplification of target genomic loci with minimal errors (e.g., Q5, KAPA HiFi). | NEB (M0491S) |
| NGS Library Prep Kit | For preparing amplified target sites for Illumina sequencing. | Illumina (TruSeq DNA PCR-Free) |
| CRISPResso2 Software | Bioinformatics tool specifically designed to quantify genome editing outcomes from NGS data. | Open Source (GitHub) |
Within the broader thesis on how adenine base editors (ABEs) function, the core catalytic transformation is the hydrolytic deamination of adenine to inosine, catalyzed by an engineered transfer RNA-specific adenosine deaminase (TadA). This whitepaper details the precise chemical mechanism, enzyme engineering trajectory, and quantitative kinetics of this foundational reaction, which enables the programmable conversion of A•T to G•C base pairs in DNA without requiring double-strand breaks.
Chemical Mechanism of Adenine Deamination
The reaction catalyzed by engineered TadA variants is a hydrolytic deamination that occurs at the C6 position of the adenine nucleobase. The mechanism proceeds via a tetrahedral intermediate.
- Activation and Nucleophilic Attack: A zinc-bound water molecule (or hydroxide ion) in the active site acts as a nucleophile, attacking the C6 carbon of the adenine ring. This is facilitated by a conserved glutamate residue (Glu59 in E. coli TadA) that acts as a general base to deprotonate the water molecule.
- Formation of Tetrahedral Intermediate: Attack at C6 leads to the formation of a tetrahedral intermediate, stabilized by coordination to the active site zinc ion.
- Ammonia Elimination: The C6-NH₂ group is protonated and eliminated as ammonia (NH₃), restoring aromaticity to the purine ring.
- Product Release: The resulting inosine product is released, completing the catalytic cycle. Inosine is subsequently read as guanosine by cellular polymerases.
Key Active Site Residues
The engineered TadA enzyme retains the essential catalytic triad from the wild-type tRNA-deaminase:
- Zinc ion (Zn²⁺): Coordinated by conserved cysteine and histidine residues (e.g., Cys106, His106, Cys149 in E. coli TadA), it polarizes the water molecule and stabilizes the transition state.
- Glu59 (E. coli numbering): Acts as a general base to activate the water nucleophile.
- Asp109: Helps orient the substrate and stabilize the transition state.
Evolution of Engineered TadA
The creation of DNA-active TadA involved extensive protein evolution. Wild-type TadA is an obligate tRNA-binding homodimer with no activity on single-stranded DNA (ssDNA).
| Evolution Phase | Key Mutations (Representative) | Functional Outcome | Catalytic Efficiency (kcat/KM) on DNA |
|---|---|---|---|
| Wild-type TadA | N/A | tRNA-specific deaminase; homodimer. | No detectable activity on ssDNA |
| TadA* (Monomer) | D108N, D147Y, E155V | Enables function as a monomer; gains low, non-specific DNA deamination activity. | ~1.3 x 10³ M⁻¹s⁻¹ |
| TadA7.10 (ABE7.10) | Additional: V106W, H123Y, S146C, D147Y, E155V, K157N | Dramatically enhanced DNA activity and specificity; used in first-generation ABEs. | ~1.1 x 10⁵ M⁻¹s⁻¹ |
| TadA8e (ABE8e) | Additional: L84F, A106V, D108N, H123Y, D147Y, E155V, K157N, R161Q | ~1000x faster than ABE7.10; tolerates non-optimal ssDNA structures. | ~1.3 x 10⁸ M⁻¹s⁻¹ |
| TadA8s (ABE8s) | A106V, D108N, H123Y, D147Y, E155V, K157N | Improved specificity profile with reduced RNA and bystander editing. | ~8.5 x 10⁷ M⁻¹s⁻¹ |
Experimental Protocol: In Vitro Deamination Kinetics Assay
This protocol measures the kinetic parameters (kcat, KM) of purified engineered TadA on ssDNA substrates.
Materials:
- Purified TadA variant (e.g., TadA8e)
- Fluorescently-labeled ssDNA oligonucleotide substrate (e.g., 5'-FAM- [Target A site] -BHQ1)
- Reaction Buffer: 20 mM HEPES pH 7.5, 150 mM KCl, 1 mM DTT, 0.1 mg/mL BSA
- Stop Solution: 95% Formamide, 10 mM EDTA
- Denaturing Urea-PAGE gel apparatus
- Fluorescence gel scanner or analyzer.
Procedure:
- Prepare Reactions: Mix 50 nM ssDNA substrate with varying concentrations of TadA enzyme (e.g., 5 nM to 5 µM) in reaction buffer at 37°C.
- Time Points: Aliquot reaction mixture at set time points (e.g., 0, 15s, 30s, 1m, 2m, 5m, 10m) into pre-prepared stop solution to halt the reaction.
- Product Separation: Heat all quenched samples to 95°C for 5 min and resolve via denaturing urea-PAGE (15% gel).
- Quantification: Use a fluorescence scanner to quantify the bands corresponding to substrate (quenched fluorescence due to proximity quenching) and product (cleaved, fluorescent).
- Kinetic Analysis: Plot initial velocity (v0) vs. enzyme concentration [E] to determine kcat. Plot v0 vs. substrate concentration [S] and fit data to the Michaelis-Menten equation to derive KM and kcat.
Visualization: ABE Architecture and Catalytic Cycle
Diagram 1: ABE Architecture & Editing Pathway
Diagram 2: TadA Catalytic Cycle
The Scientist's Toolkit: Key Reagents for ABE Research
| Reagent/Solution | Function & Description |
|---|---|
| Engineered TadA Plasmid (e.g., pCMV_ABE8e) | Mammalian expression vector encoding the TadA-nCas9 fusion protein under a CMV promoter. Essential for delivery into cells. |
| sgRNA Expression Plasmid (e.g., pU6-sgRNA) | Vector for expressing single-guide RNA targeting the genomic locus of interest under a U6 promoter. |
| HEK293T Cells | A robust, easily transfected human cell line commonly used for initial validation of base editing efficiency and specificity. |
| Lipofectamine 3000 | A cationic lipid-based transfection reagent for delivering plasmid DNA into mammalian cells. |
| Next-Generation Sequencing (NGS) Library Prep Kit (e.g., Illumina) | For deep sequencing of the target locus to quantify base editing efficiency (%), indel rate, and analyze bystander/edit product distribution. |
| Purified TadA Protein (e.g., TadA8e) | Recombinant protein for in vitro biochemical assays to study kinetics, substrate specificity, and structural biology (e.g., crystallography). |
| Fluorescent ssDNA Substrate | Oligonucleotide with a target adenine, used in fluorescence-quenching or gel-based in vitro deamination assays to measure enzyme activity. |
| Ribonucleoprotein (RNP) Complex | Pre-assembled complex of purified nCas9-TadA protein and synthetic sgRNA. Used for direct delivery (e.g., electroporation) to minimize off-target effects and for ex vivo therapeutic applications. |
| BEAT (Base Editing Analytical Tool) | Bioinformatics software for analyzing NGS data to calculate precise base editing outcomes from sequencing reads. |
Within the broader thesis on "How do adenine base editors (ABEs) work?", understanding the evolution of the protein architecture is paramount. Adenine Base Editors enable the direct, programmable conversion of adenine (A) to guanine (G) in DNA without inducing double-strand breaks. This technical guide details the core generations, from the pioneering ABE7.10 to modern high-efficiency, high-fidelity variants, framing their development within the context of mechanistic optimization for research and therapeutic applications.
Mechanistic Foundation of ABE Function
ABEs function via a fusion protein consisting of a catalytically impaired Cas9 nickase (nCas9) and an engineered adenine deaminase enzyme. The nCas9 targets the complex to a specific genomic locus via a guide RNA (gRNA). Within the resultant single-stranded DNA R-loop, the deaminase catalyzes the hydrolytic deamination of adenine to form inosine, which is read as guanine by DNA polymerases. Subsequent cellular DNA repair mechanisms, initiated by the nick in the non-edited strand, permanently install the A•T to G•C base pair change.
Diagram Title: ABE Core Catalytic Mechanism
Generational Evolution: A Technical Breakdown
First Generation: ABE7.10
ABE7.10, evolved from E. coli TadA, demonstrated the first proof-of-concept for A-to-G editing in mammalian cells. Its efficiency was moderate, and it exhibited sequence context preferences (e.g., optimal activity in a YAC motif, where Y = C/T).
Key Optimized Variants
Subsequent generations focused on improving efficiency, product purity (reducing indels and byproducts), broadening sequence compatibility, and improving delivery.
| Editor Name | Key Modifications | Primary Improvement | Typical Editing Efficiency Range* | Product Purity (A-to-G) |
|---|---|---|---|---|
| ABE7.10 | TadA7.10 dimer (wtTadA + TadA7.10) | Proof-of-concept | 10-50% | Moderate |
| ABE8e | 8 further mutations in TadA*8e | Enhanced activity & rate | 50-80% | High |
| ABE8s | 8 mutations (different set than 8e) | Reduced off-target RNA editing | 40-75% | Very High |
| ABE8a | TadA*8a variant | Broader sequence context (relaxed YAC) | 45-78% | High |
| ABE8.17-m | Dual TadA*8.17 monomers | High fidelity, reduced off-target DNA editing | 30-70% | Extremely High |
| ABE9e | Engineered TadA-Nme2 variant | Ultra-high product purity, minimal Cas9-independent off-target | 20-60% | Near Maximum |
| ABEmax | Codon/context optimization | Improved expression in mammalian cells | Comparable to parent variant | Comparable to parent variant |
*Efficiency is highly dependent on target site, cell type, and delivery method. Ranges are illustrative from literature.
Experimental Protocol: Evaluating ABE Variants in HEK293T Cells
Objective: Compare on-target editing efficiency and product purity of two ABE variants at multiple genomic loci.
- gRNA Design & Cloning: Design 3-5 gRNAs targeting distinct genomic sites with varying sequence contexts. Clone gRNA sequences into an appropriate plasmid backbone (e.g., pCMVABE#U6-gRNA).
- Cell Transfection: Seed HEK293T cells in 24-well plates. At 70-80% confluency, co-transfect 500 ng of ABE expression plasmid (e.g., ABE8e vs. ABE8.17-m) and 250 ng of the respective gRNA plasmid per well using a polyethylenimine (PEI) protocol.
- Harvest Genomic DNA: 72 hours post-transfection, harvest cells and extract genomic DNA using a silica-column-based kit.
- PCR Amplification: Amplify target loci (500-700 bp amplicons) using high-fidelity PCR.
- Next-Generation Sequencing (NGS) Library Prep: Purify PCR products and prepare sequencing libraries using a dual-indexing amplicon sequencing kit. Pool libraries.
- NGS & Data Analysis: Sequence on an Illumina MiSeq. Process reads using a base-editing analysis pipeline (e.g, BEAT or CRISPResso2) to calculate:
- Editing Efficiency: (% of reads with A-to-G conversion at the target base).
- Product Purity: (% of edited reads containing only the desired A-to-G change, without other substitutions or indels).
- Byproduct Analysis: Frequency of other base substitutions (e.g., A-to-C/T) and indel formation.
The Scientist's Toolkit: Essential Research Reagents
| Reagent / Material | Function in ABE Research | Example Vendor/ID |
|---|---|---|
| ABE Expression Plasmids | Mammalian expression vector for the ABE fusion protein (nCas9-TadA). | Addgene (#112101 for ABE7.10, #138489 for ABE8e) |
| gRNA Cloning Vector | Plasmid with U6 promoter for expression of single guide RNA (sgRNA). | Addgene (#41824) |
| PEI Transfection Reagent | Polycationic polymer for efficient plasmid delivery into mammalian cells. | Polysciences, linear PEI 25k |
| Genomic DNA Extraction Kit | Silica-membrane based kit for high-quality, PCR-ready DNA. | Qiagen DNeasy Blood & Tissue Kit |
| High-Fidelity PCR Master Mix | Enzyme mix for accurate amplification of target loci for sequencing. | NEB Q5 Hot Start Master Mix |
| Amplicon NGS Library Prep Kit | Kit for attaching Illumina sequencing adapters and indices to PCR amplicons. | Illumina DNA Prep Kit |
| HEK293T Cell Line | Robust, easily transfected human cell line for initial base editor characterization. | ATCC CRL-3216 |
| Sanger Sequencing Service | For rapid, low-throughput validation of editing outcomes. | Eurofins Genomics |
| BE Analysis Software | Computational tool to quantify base editing outcomes from NGS data. | CRISPResso2 (Broad Institute) |
Diagram Title: ABE On-Target Evaluation Workflow
Advanced Considerations & Pathway to Therapy
The evolution of ABEs involves balancing multiple molecular pathways. High-efficiency variants like ABE8e accelerate the deamination pathway, but may increase off-target effects. High-fidelity variants like ABE8.17-m and ABE9e introduce mutations that minimize off-target deamination pathways while retaining robust on-target activity, a crucial balance for therapeutics.
Diagram Title: ABE Evolution Drivers and Solutions
The trajectory from ABE7.10 to high-efficiency, high-fidelity variants like ABE8e and ABE9e represents a deliberate engineering effort to refine the mechanistic action of the adenine deaminase-Cas9 fusion. This evolution, central to the thesis on how ABEs work, has transitioned the technology from a novel proof-of-concept to a precise and programmable tool capable of correcting a majority of known pathogenic point mutations for research and clinical drug development. Future generations will continue to optimize the interplay between efficiency, specificity, and delivery.
Base editing, particularly Adenine Base Editor (ABE) technology, represents a precise method for genome engineering, enabling the direct, irreversible conversion of adenine (A) to guanine (G) without inducing double-strand DNA breaks. Within the broader thesis on "How do adenine base editors (ABEs) work?", a critical operational parameter is the editing window—the narrow region within a target DNA site where efficient base conversion occurs. This window is fundamentally constrained by two interdependent factors: the spatial positioning of the protospacer sequence and the requisite presence of a Protospacer Adjacent Motif (PAM). This guide dissects these requirements and their quantitative impact on editing efficiency and precision.
Protospacer Positioning and the Catalytic Window
The ABE fusion protein consists of a catalytically impaired Cas9 nickase (most commonly nSpCas9) tethered to an engineered adenine deaminase enzyme (e.g., TadA-8e). The deaminase domain has a fixed spatial relationship to the Cas9 protein. Consequently, only adenines located within a specific span of nucleotides opposite the deaminase, typically positions 4 to 8 (counting the PAM as positions 21-23), are accessible for conversion. This defines the catalytic or editing window.
Table 1: Editing Window Characteristics for Common ABE Variants
| ABE Variant | Cas9 Scaffold | Primary Editing Window (A positions) | Deaminase Domain | Key Reference |
|---|---|---|---|---|
| ABE7.10 | nSpCas9 | A4-A8 | TadA-7.10 | Gaudelli et al., 2017 |
| ABE8e | nSpCas9 | A4-A8 (with broader activity) | TadA-8e | Richter et al., 2020 |
| ABE8s | nSaCas9 | A3-A7 | TadA-8e | Richter et al., 2020 |
| ABEmax | nSpCas9 | A4-A8 | TadA-8e | Koblan et al., 2018 |
Diagram: ABE Architecture and Catalytic Window
PAM Requirements and Compatibility
The PAM sequence is essential for Cas9 recognition and binding. The canonical PAM for Streptococcus pyogenes Cas9 (SpCas9) is 5'-NGG-3', where 'N' is any nucleotide. This requirement directly dictates which genomic loci can be targeted and determines the orientation and distance of the editing window relative to the PAM. The development of engineered Cas9 variants with altered PAM specificities (e.g., SpG, SpRY, xCas9) has significantly expanded the targetable space for ABEs.
Table 2: PAM Compatibility and Target Range for Cas9 Variants used in ABEs
| Cas9 Variant | PAM Requirement | Canonical Example | Approx. Targeting Density (in human genome) | Use in ABE |
|---|---|---|---|---|
| SpCas9 | NGG | 5'-AGG-3' | ~1 in 16 bp | Standard (ABE7.10, ABEmax) |
| SpCas9-NG | NG | 5'-GAC-3' | ~1 in 4 bp | ABE8e-NG |
| SpRY | NRN > NYN | 5'-NAC-3' | Nearly PAM-less | ABE8e-SpRY |
| SaCas9 | NNGRRT | 5'-GAGACC-3' | ~1 in 32 bp | ABE8s |
| SaCas9-KKH | NNNRRT | 5'-CGAAGT-3' | ~1 in 16 bp | ABE8.8e-KKH |
Experimental Protocol: Determining Editing Window and Efficiency
Objective: To empirically map the editing window and efficiency profile of an ABE variant at a defined genomic locus.
Methodology:
- Target Selection and sgRNA Design: Select a genomic locus containing multiple adenines (As) within the putative editing window (e.g., positions 1-20 of the protospacer). Design an sgRNA complementary to the target protospacer sequence adjacent to a compatible PAM.
- Cell Transfection: Transfect cultured mammalian cells (e.g., HEK293T) with two plasmids: one expressing the ABE variant and the other expressing the designed sgRNA. Include a negative control (sgRNA only).
- Harvest and Genomic DNA Extraction: Harvest cells 72-96 hours post-transfection. Extract genomic DNA using a commercial kit.
- PCR Amplification: Amplify the target genomic region using high-fidelity PCR primers flanking the edited site.
- Next-Generation Sequencing (NGS) Analysis: Purify PCR amplicons and prepare an NGS library. Perform deep sequencing (≥10,000x coverage per sample).
- Data Analysis: Align sequences to the reference genome. Calculate the A-to-G editing efficiency at each adenine position within the protospacer as:
(Number of G reads / Total reads at that position) × 100%. Plot efficiency against adenine position to visualize the editing window.
Diagram: Experimental Workflow for Editing Window Analysis
Table 3: Example NGS Data from an ABE8e Experiment (Hypothetical Data)
| Adenine Position (relative to PAM) | Sequence Context (5'→3') | Total Reads | Edited Reads (G) | Editing Efficiency (%) |
|---|---|---|---|---|
| A1 | ACTAGCT...GGT | 12500 | 12 | 0.1 |
| A2 | ACATGCT...GGT | 11800 | 24 | 0.2 |
| A3 | AACTGCG...GGT | 12200 | 122 | 1.0 |
| A4 | AACTGCG...GGT | 12050 | 3013 | 25.0 |
| A5 | ACTGACG...GGT | 11980 | 4792 | 40.0 |
| A6 | ACTGCAG...GGT | 12300 | 4305 | 35.0 |
| A7 | CTGCAAT...GGT | 11700 | 2340 | 20.0 |
| A8 | TGCAAAC...GGT | 12100 | 1210 | 10.0 |
| A9 | GCAAAAT...GGT | 12000 | 120 | 1.0 |
| A10 | CAAATAA...GGT | 11900 | 0 | 0.0 |
The Scientist's Toolkit: Key Research Reagents
| Item | Function/Description | Example Product/Catalog |
|---|---|---|
| ABE Expression Plasmid | Mammalian expression vector for the ABE fusion protein (e.g., ABE8e). | Addgene #138489 (pCMV_ABE8e) |
| sgRNA Expression Plasmid | U6-promoter driven vector for sgRNA cloning and expression. | Addgene #41824 (pU6-sgRNA) |
| High-Efficiency Transfection Reagent | For delivering plasmids into mammalian cells. | Lipofectamine 3000, PEIpro |
| Genomic DNA Extraction Kit | For rapid, pure gDNA isolation from mammalian cells. | Qiagen DNeasy Blood & Tissue Kit |
| High-Fidelity PCR Polymerase | For accurate amplification of the target locus for NGS. | Q5 High-Fidelity DNA Polymerase (NEB) |
| NGS Library Prep Kit for Amplicons | For preparing barcoded Illumina sequencing libraries from PCR products. | Illumina DNA Prep Kit |
| HEK293T Cell Line | A robust, easily transfected human cell line for initial ABE validation. | ATCC CRL-3216 |
| Sanger Sequencing Service | For initial, rapid verification of editing. | In-house or commercial providers |
How to Use ABEs: Protocols, Design, and Cutting-Edge Applications in Biomedicine
Adenine Base Editors (ABEs) are precision genome editing tools that catalyze the conversion of A•T base pairs to G•C without inducing double-stranded DNA breaks. Within the broader thesis on How do adenine base editors (ABEs) work?, a critical determinant of experimental success is the design of the single guide RNA (sgRNA). This guide provides a technical protocol for designing sgRNAs to maximize ABE efficiency and purity.
Principles of ABE Activity and sgRNA Design
ABEs are fusion proteins consisting of a catalytically impaired Cas9 nickase (nCas9) fused to an engineered adenine deaminase enzyme. The sgRNA directs the ABE complex to the target genomic locus, where the deaminase acts on a single-stranded DNA window exposed by the Cas9-sgRNA complex. Optimal sgRNA design must consider editing window placement, sequence context, and off-target avoidance.
Key Quantitative Parameters for ABE sgRNA Design:
- Editing Window: Positions 4-8 (most efficient) within the protospacer, relative to the PAM (NGG, located at positions 21-23).
- Sequence Context: The deaminase has preferences for certain local sequence motifs.
- Strand Selection: ABEs primarily deaminate adenines on the non-complementary (displaced) strand of the DNA bubble.
Diagram Title: ABE Mechanism and sgRNA-Dependent Target Strand Displacement
Step-by-Step sgRNA Design Protocol
Step 1: Define Target Adenine and PAM Orientation
Identify the specific adenine (A) residue(s) you intend to convert. The effective editing window for ABE8e (a common high-activity variant) is typically positions 4-8 (1-indexed) within the 20-nt protospacer, 5' of the NGG PAM. Therefore, the target A must be positioned accordingly.
- For a target A on the top strand, the NGG PAM will be located 3' (downstream) of the target sequence.
- For a target A on the bottom strand, the NGG PAM will be located 5' (upstream) of the target sequence.
Step 2: Generate Candidate sgRNA Sequences
For each target A, design two candidate sgRNAs:
- sgRNA (Target A on Top Strand): 20-nt sequence directly upstream of an NGG PAM.
- sgRNA (Target A on Bottom Strand): 20-nt sequence directly downstream of an NGG PAM (you will use the reverse complement as the sgRNA).
Rule: The target adenine should ideally fall at positions A4-A8 within the protospacer.
Step 3: Apply Sequence Context Filters
Prioritize sgRNAs where the target adenine is within a favorable sequence context. Research indicates preferences, though these can vary by ABE variant.
Table 1: Sequence Context Preferences for ABE Deamination
| Sequence Motif (5' to 3') | Relative Efficiency | Notes |
|---|---|---|
| T/A/C-A-C/T/A | High | Contexts like TAC, CAC often show robust activity. |
| G-A-A/G | Moderate | Can be efficient but may vary. |
| G-A-C/G | Lower | May require high-activity ABE variants (e.g., ABE8e). |
| Poly-A Tracts | Avoid | Consecutive As can lead to bystander edits. |
Step 4: Minimize Off-Target Predictions
Use standard CRISPR/Cas9 off-target prediction tools (e.g., Cas-OFFinder, CRISPOR) to screen your candidate sgRNA sequences. Filter out sgRNAs with perfect or near-perfect matches (≤3 mismatches) elsewhere in the genome, especially in coding regions.
Step 5: Optimize for Specificity & Purity (Avoid Bystander Edits)
If your target "A" is within a stretch of multiple adenines, neighboring "A"s within the editing window may also be deaminated, creating unwanted bystander mutations.
- Strategy: Re-orient your sgRNA design (shift the PAM location) so that only your intended A falls within positions 4-8, and other nearby As are positioned outside this window (e.g., at position 3 or 9+).
Step 6: Final Selection and Synthesis
Select 2-3 top-ranking sgRNAs per target for empirical testing. Order as synthetic crRNA (for Cas9 systems using tractRNA) or as full-length sgRNA, with appropriate chemical modifications (e.g., 2'-O-methyl 3' phosphorothioate) to enhance stability.
Experimental Validation Protocol
Title: In vitro Validation of ABE sgRNA Activity Using a HEK293T Reporter Cell Line
Objective: To quantitatively compare the editing efficiency and purity of candidate sgRNAs.
Materials:
- Reporter Plasmid: Contains a disrupted GFP gene, restored only by a specific A•T to G•C conversion at the target site.
- ABE Expression Plasmid: e.g., pCMV_ABE8e.
- sgRNA Expression Plasmids: U6-promoter driven constructs for each candidate.
- Cells: HEK293T cells.
- Transfection Reagent: PEI or lipofectamine-based.
- Flow Cytometer.
Procedure:
- Seed HEK293T cells in a 24-well plate.
- Co-transfect cells with:
- 500 ng ABE expression plasmid.
- 250 ng sgRNA expression plasmid.
- 250 ng GFP reporter plasmid.
- Include controls (No-ABE, No-sgRNA).
- Incubate cells for 72 hours.
- Harvest cells and analyze the percentage of GFP-positive cells via flow cytometry.
- Calculate editing efficiency: % GFP+ (sample) - % GFP+ (No-ABE control).
- For leads, isolate genomic DNA and perform targeted next-generation sequencing (NGS) to assess precise edit percentage and bystander edit profile.
Table 2: Example sgRNA Validation Data (Hypothetical)
| sgRNA ID | Target A Position | Sequence Context | Flow Cytometry %GFP+ | NGS % Intended Edit | NGS % Bystander Edits (A5) |
|---|---|---|---|---|---|
| sgRNA-1 | A5 | TAC | 42% | 38% | <1% |
| sgRNA-2 | A6 | GAC | 28% | 22% | 15% |
| sgRNA-3 | A8 | CAC | 35% | 31% | <1% |
Diagram Title: Experimental Workflow for sgRNA Validation
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for ABE sgRNA Design & Testing
| Reagent / Solution | Function / Purpose | Example Product / Vendor |
|---|---|---|
| ABE Expression Plasmid | Delivers the base editor protein (e.g., ABE8e) into cells. | pCMV_ABE8e (Addgene #138489) |
| sgRNA Cloning Vector | Backbone for expressing sgRNA from a U6 promoter. | pU6-sgRNA (Addgene #136833) |
| Reporter Plasmid | Enables rapid, quantitative assessment of base editing efficiency via fluorescence or other reporters. | HEK293T GFP-based ABE reporter (e.g., Addgene # DNA repair reporter lines) |
| NGS Amplicon-Seq Kit | For preparing sequencing libraries from PCR-amplified target genomic loci to quantify editing outcomes. | Illumina DNA Prep, or IDT xGen Amplicon kits |
| Off-Target Prediction Tool | In silico assessment of potential off-target sites for a given sgRNA sequence. | Cas-OFFinder (webbased), CRISPOR (webbased) |
| Chemically Modified sgRNA | Synthetic sgRNA with enhanced nuclease resistance and stability for direct delivery (e.g., RNP). | Synthego V2 sgRNA (2'-O-methyl, phosphorothioate) |
| Genomic DNA Isolation Kit | High-quality DNA extraction for downstream NGS analysis post-editing. | Qiagen DNeasy Blood & Tissue Kit |
| Flow Cytometry Assay Kit | For analyzing fluorescent reporter restoration in live cells. | Not applicable; requires flow cytometer instrument. |
This whitepaper explores the core delivery systems enabling the revolutionary technology of Adenine Base Editors (ABEs). ABEs, which catalyze the direct, irreversible conversion of adenine (A) to guanine (G) in genomic DNA without causing double-strand breaks, require efficient and precise delivery into target cells. The choice of delivery vehicle—from plasmid DNA to viral vectors and ribonucleoprotein (RNP) complexes—profoundly impacts editing efficiency, specificity, translational safety, and therapeutic applicability. This guide provides an in-depth technical comparison of these systems within the context of ABE research.
Core Delivery Modalities for ABEs
Plasmid Transfection
Plasmid-based delivery involves introducing circular DNA encoding the ABE machinery (e.g., ABE8e) and, optionally, a single-guide RNA (sgRNA) into cells.
- Mechanism: The plasmid must enter the nucleus for transcription and translation of the ABE protein and sgRNA (if expressed).
- Advantages: Simple, low-cost production; large cargo capacity.
- Disadvantages: Low efficiency in primary/non-dividing cells; risk of genomic integration and long-term expression leading to off-target effects; high immunogenicity.
Viral Vectors
Viral vectors are engineered to deliver genetic material encoding ABEs while being replication-incompetent.
Table 1: Key Viral Vectors for ABE Delivery
| Vector Type | Packaging Capacity | Tropism | Integration | Pros for ABE Delivery | Cons for ABE Delivery |
|---|---|---|---|---|---|
| Adeno-Associated Virus (AAV) | ~4.7 kb | Broad (serotype-dependent) | Non-integrating (episomal) | High in vivo delivery efficiency; low immunogenicity; strong safety profile. | Limited capacity requires split-ABE systems; potential for long-term expression. |
| Lentivirus (LV) | ~8 kb | Broad (including non-dividing cells) | Integrating (random) | High efficiency in hard-to-transfect cells; stable expression. | Risk of insertional mutagenesis; long-term expression increases off-target potential. |
| Adenovirus (AdV) | ~8-36 kb | Broad | Non-integrating | Very high transduction efficiency; large capacity. | High immunogenicity limits re-dosing; complex production. |
Ribonucleoprotein (RNP) Delivery
RNP delivery involves the direct introduction of pre-assembled complexes of purified ABE protein and in vitro-transcribed sgRNA.
- Mechanism: The RNP complex is directly active upon cellular entry and rapidly degrades, minimizing its window of activity.
- Advantages: Rapid editing with minimal off-target effects; no risk of genomic integration; no immunogenicity from foreign DNA/RNA.
- Disadvantages: Lower delivery efficiency requiring physical methods (e.g., electroporation); transient activity unsuitable for some in vivo applications; complex manufacturing.
Table 2: Quantitative Comparison of ABE Delivery Systems
| Parameter | Plasmid Transfection | AAV Vector | Lentiviral Vector | RNP Delivery |
|---|---|---|---|---|
| Typical Editing Efficiency (in vitro) | 5-30% (cell line dependent) | 20-80% (in vivo & in vitro) | 40-90% (in vitro) | 40-95% (in primary cells via electroporation) |
| Time to Peak Editing | 24-72 hours | 3-14 days | 3-7 days | < 24 hours |
| Expression Kinetics | Transient to stable (if integrated) | Prolonged (weeks-months) | Stable (integrated) | Transient (hours) |
| Risk of Off-Target Effects | High (persistent expression) | Moderate (prolonged expression) | High (stable integration) | Low (transient activity) |
| Cargo Flexibility | High (unlimited size) | Low (requires splitting ABE) | Moderate | High (protein can be engineered) |
| Suitability for In Vivo Therapy | Low | High | Moderate (safety concerns) | Emerging (local delivery) |
Detailed Experimental Protocols for Key ABE Delivery Methods
Protocol 1: ABE8e RNP Delivery via Nucleofection into Primary Human T Cells
This protocol is critical for ex vivo therapeutic applications like CAR-T cell engineering.
Materials (Research Reagent Solutions):
- Cells: Isolated primary human CD4+/CD8+ T cells.
- ABE Protein: Purified ABE8e-NLS protein (commercially available or expressed from E. coli).
- sgRNA: Chemically modified sgRNA targeting the TRAC locus, resuspended in nuclease-free buffer.
- Nucleofector Kit: P3 Primary Cell 4D-Nucleofector X Kit (Lonza).
- Equipment: 4D-Nucleofector System, 37°C CO2 incubator.
Procedure:
- RNP Complex Formation: Incubate ABE8e protein (60 pmol) and sgRNA (120 pmol) at room temperature for 10 minutes in a small volume of PBS.
- Cell Preparation: Isolate and count T cells. Centrifuge 1-2e6 cells, aspirate supernatant completely.
- Nucleofection: Resuspend cell pellet in 100 µL of pre-warmed P3 Nucleofector Solution. Mix with the pre-formed RNP complex. Transfer to a Nucleocuvette and electroporate using pulse code EO-115.
- Recovery: Immediately add 500 µL of pre-warmed culture medium to the cuvette. Transfer cells to a pre-warmed 24-well plate. Incubate at 37°C, 5% CO2.
- Analysis: Harvest cells 48-72 hours post-nucleofection for genomic DNA extraction. Assess editing efficiency via targeted next-generation sequencing (NGS) or RFLP assay.
Protocol 2: In Vivo ABE Delivery using AAV9 for Liver Targeting
This protocol is standard for creating disease models or for therapeutic liver editing.
Materials (Research Reagent Solutions):
- AAV Vectors: AAV9 vectors encoding ABE8e (split as N-terminal and C-terminal halves, SaCas9 or smaller Cas variant) and the matching sgRNA under U6 promoter.
- Animal Model: 6-8 week old C57BL/6 mice.
- Reagents: PBS for dilution.
- Equipment: Insulin syringes, animal restraint device.
Procedure:
- Vector Preparation: Thaw AAV-ABE and AAV-sgRNA stocks on ice. Mix vectors at an appropriate titer (e.g., 1e11 vg each per mouse) in PBS for a final injection volume of 100-200 µL.
- Administration: Restrain the mouse and perform a tail vein injection slowly and steadily.
- Monitoring: House injected mice for 4-8 weeks to allow for robust transgene expression and editing.
- Tissue Harvest & Analysis: Euthanize mice and harvest liver tissue. Homogenize and extract genomic DNA. Quantify editing efficiency at the target locus via NGS. Assess potential off-target editing at predicted sites.
Visualizations
Title: Plasmid Transfection Workflow for ABE Delivery
Title: Viral Vector Pathways: AAV vs Lentivirus
Title: RNP Delivery Mechanism for ABEs
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for ABE Delivery Experiments
| Item | Function in ABE Delivery | Example/Note |
|---|---|---|
| ABE Expression Plasmid | Source of ABE and sgRNA genes for plasmid transfection or viral vector production. | pCMV-ABE8e, often requires a separate U6-sgRNA plasmid. |
| AAV Helper & Rep/Cap Plasmid | For AAV vector production; provides replication and capsid proteins in trans. | pHelper, pAAV-RC9 (for serotype 9). |
| Lentiviral Packaging Mix | Plasmid mix (gag/pol, rev, VSV-G) for producing replication-incompetent lentivirus. | Third-generation systems for enhanced safety. |
| Purified ABE Protein | Recombinant base editor protein for RNP assembly. Must contain Nuclear Localization Signals (NLS). | ABE8e (NLS-tagged), commercial or in-house purified. |
| Chemically Modified sgRNA | Enhanced stability and reduced immunogenicity for RNP or viral delivery. | Chemical modifications (e.g., 2'-O-methyl, phosphorothioate) at terminal nucleotides. |
| Lipofectamine 3000 | Cationic lipid reagent for plasmid transfection into adherent cell lines. | Standard for in vitro efficiency testing. |
| Nucleofector Kit | Electroporation-based system for high-efficiency RNP delivery into hard-to-transfect cells. | Cell-type specific kits (e.g., Lonza P3, Amaxa). |
| Polybrene / Protamine Sulfate | Cationic polymers that enhance viral transduction efficiency. | Used during lentiviral spinoculation. |
| DNase I | Essential for degrading unpackaged DNA during AAV or LV purification (clarified lysate). | Ensures accurate viral titer measurement. |
| Anti-AAV Neutralizing Antibody Assay | To screen for pre-existing immunity against specific AAV serotypes in subjects. | Critical for planning in vivo studies/therapies. |
The advancement of ABE therapeutics is inextricably linked to progress in delivery technology. Plasmid methods remain useful for initial in vitro screening, while viral vectors, particularly AAV, are indispensable for efficient in vivo delivery. However, the RNP platform is emerging as the gold standard for applications demanding maximal precision and safety, especially in ex vivo settings. The choice of system involves a critical trade-off between editing efficiency, persistence, specificity, and immunogenicity. Future developments in vector engineering (e.g., engineered capsids, non-viral nanoparticles) and RNP delivery optimization will further unlock the full clinical potential of adenine base editing.
This whitepaper is framed within the broader thesis investigating How do adenine base editors (ABEs) work?. A core pillar of this research involves applying the mechanistic understanding of ABE function—a fusion of a catalytically impaired Cas9 nickase (nCas9) and an engineered adenine deaminase—to diverse biological models. The transition from in vitro characterization to sophisticated in vitro, ex vivo, and in vivo model systems is critical for validating editing efficiency, specificity, and functional outcomes. This guide details the technical application of ABEs across three foundational tiers: immortalized cell lines, patient-derived organoids, and animal models, providing a roadmap for researchers to design rigorous experiments.
Table 1: Comparative Performance of ABE Systems Across Model Systems
| Model System | Typical ABE Construct | Average Editing Efficiency Range (%) | Primary Readout Method | Key Advantage | Major Limitation |
|---|---|---|---|---|---|
| Immortalized Cell Lines (e.g., HEK293T, HeLa) | ABEmax, ABE8e | 30-80% | NGS, Sanger Sequencing, Phenotypic Assay | High-throughput, reproducible, easy to culture | Lack of native genomic context & physiology |
| Patient-Derived Organoids (e.g., intestinal, hepatic) | ABE7.10, ABEmax | 10-50% | NGS, Immunofluorescence, Functional Assay | Captures patient genetics, 3D architecture, some physiology | Heterogeneous editing, costly, time-consuming |
| Mouse Models (In Vivo) | AAV-delivered ABE8e | 5-40% (tissue-dependent) | NGS, IHC, Behavioral/Biochemical Assay | Systemic or tissue-specific delivery, whole-organism physiology | Lower efficiency, potential for immune response, off-target concerns |
Table 2: Common Delivery Methods and Specifications
| Model System | Preferred Delivery Method | Vector Type | Typical Transfection/Infection Efficiency | Key Considerations |
|---|---|---|---|---|
| Cell Lines | Lipid-based Transfection | Plasmid DNA | 70-95% | Optimize lipid:DNA ratio; use split-intein systems for larger constructs. |
| Organoids | Electroporation or Lentivirus | RNP or LV | 20-60% | Nucleofection protocols are organoid-specific; LV allows stable expression. |
| In Vivo (Mouse) | Adeno-Associated Virus (AAV) | AAV9, AAV-DJ | Varies by serotype & tissue | Dose critical; AAV cargo size limit (~4.7kb) necessitates compact editors. |
Experimental Protocols for Key Applications
Protocol 3.1: ABE Editing in HEK293T Cell Lines for Functional Validation
Objective: Introduce a precise A•T to G•C point mutation in a gene of interest (GOI) to study functional consequences.
- Design and Cloning: Design sgRNA targeting the adenine within the editing window (typically positions 4-8, protospacer-adjacent motif [PAM] distal). Clone sgRNA into an ABE expression plasmid (e.g., pCMV_ABEmax).
- Cell Culture and Transfection: Maintain HEK293T cells in DMEM + 10% FBS. Seed 2.5e5 cells/well in a 12-well plate 24h pre-transfection. Transfect with 1 µg ABE plasmid + 0.5 µg sgRNA plasmid using polyethylenimine (PEI) at a 3:1 (PEI:DNA) ratio.
- Harvest and Analysis: Harvest cells 72h post-transfection. Extract genomic DNA.
- Assessment:
- Efficiency: Amplify target locus by PCR. Submit for Sanger sequencing and analyze trace decomposition (e.g., with EditR or BEAT) or perform high-throughput NGS.
- Function: Perform Western blot, ELISA, or a relevant phenotypic assay (e.g., proliferation, differentiation) 5-7 days post-transfection.
Protocol 3.2: ABE Editing in Intestinal Organoids via Nucleofection
Objective: Correct a disease-associated SNP in patient-derived colon organoids.
- Organoid Culture: Maintain human intestinal organoids in Matrigel domes with Wnt/R-spondin/Noggin-enriched medium. Passage every 7-10 days.
- RNP Complex Formation: Chemically synthesize or in vitro transcribe sgRNA. Purify ABE protein (e.g., ABE8e-NG). Mix 6 µg ABE protein with 3 µg sgRNA to form ribonucleoprotein (RNP) complexes.
- Nucleofection: Dissociate organoids into single cells. Resuspend 2e5 cells in nucleofection solution (P3 Primary Cell Kit, Lonza) with RNP complexes. Electroporate using a 4D-Nucleofector (program: CM-137). Immediately recover cells in warm culture medium + 10 µM Y-27632 (ROCKi).
- Analysis: Re-embed cells in Matrigel. After 5-7 days, harvest organoids. Extract genomic DNA for NGS analysis of on-target editing and off-target screening (e.g., GUIDE-seq or targeted NGS). Confirm correction via immunohistochemistry for restored protein expression.
Protocol 3.3: In Vivo ABE Delivery via AAV in a Mouse Model
Objective: Perform targeted base editing in the mouse liver to model a metabolic disorder correction.
- Construct and Vector Preparation: Subclone a compact ABE variant (e.g., saABE8e) and liver-specific sgRNA into a single AAV vector under a liver-specific promoter (e.g., TBG). Package into AAV9 or AAV-DJ capsids via triple transfection in HEK293 cells. Purify and titer the virus.
- Animal Injection: Inject 6-8 week old mice intravenously via the tail vein with 1e11 to 5e11 vector genomes (vg) of AAV-ABE per mouse in 100 µL sterile saline.
- Tissue Collection and Processing: Euthanize mice 4-8 weeks post-injection. Perfuse with PBS, harvest liver and other potential off-target organs (e.g., heart, brain). Snap-freeze for DNA/RNA or fix for histology.
- Analysis: Extract genomic DNA from liver sections. Amplify the target region for NGS to quantify editing efficiency and specificity. Analyze serum biomarkers for phenotypic correction. Perform whole-transcriptome RNA-seq to assess unintended consequences.
Visualizations
Diagram 1: ABE Mechanism in a Cell
Diagram 2: Workflow for ABE Application Across Models
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagent Solutions for ABE Experiments
| Reagent / Material | Primary Function | Example Product / Note |
|---|---|---|
| ABE Expression Plasmid | Encodes the base editor fusion protein. Enables transient or stable expression. | pCMV_ABE8e (Addgene #138495); choose promoter (CMV, EF1α) suitable for model. |
| sgRNA Cloning Vector | Backbone for expressing sgRNA with appropriate RNA Polymerase III promoter (U6, H1). | pU6-sgRNA (Addgene #138495) or ordered as a gBlock for RNP assembly. |
| Lipid Transfection Reagent | Forms complexes with plasmid DNA for efficient delivery into cell lines. | Lipofectamine 3000 (Thermo Fisher), PEI MAX (Polysciences). |
| Nucleofection Kit | Electroporation solution optimized for difficult-to-transfect cells (e.g., organoid cells). | P3 Primary Cell 4D-Nucleofector Kit (Lonza). |
| Recombinant ABE Protein | Purified protein for RNP formation. Reduces off-targets and enables rapid editing. | Custom purified ABE8e (e.g., from Arcila, Thermo). Requires cold chain. |
| AAV Serotype (e.g., AAV9) | Viral vector for efficient in vivo delivery to specific tissues (liver, CNS, muscle). | Packaged AAV-ABE construct; titer critical for dosing. |
| Next-Generation Sequencing Kit | For deep sequencing of target loci to quantify editing efficiency and off-target effects. | Illumina MiSeq, AmpliSeq; use dual-indexed amplicon sequencing. |
| EditR Analysis Software | Python-based tool for quantifying base editing efficiency from Sanger sequencing traces. | Publicly available on GitHub; requires input of sgRNA sequence. |
| Matrigel / BME | Basement membrane extract for 3D organoid culture and post-nucleofection recovery. | Corning Matrigel, Growth Factor Reduced; keep on ice. |
| Y-27632 (ROCKi) | Rho kinase inhibitor. Reduces apoptosis in dissociated organoid cells post-nucleofection. | Add to medium at 10 µM for 24-48h after editing. |
The development of adenine base editors (ABEs) represents a pivotal advancement in the broader thesis of precision genome engineering for therapeutic intervention. Unlike traditional CRISPR-Cas9 systems that create double-strand breaks (DSBs), ABEs catalyze the direct, irreversible conversion of adenine (A) to guanine (G) in DNA without inducing DSBs. This enables the precise correction of A•T to G•C point mutations, which constitute approximately 47% of all known pathogenic human single-nucleotide variants (SNVs). This technical guide details the application of ABEs for correcting such mutations, covering mechanistic principles, experimental protocols, and key research tools.
Mechanism of Adenine Base Editors (ABEs)
ABEs are fusion proteins consisting of a catalytically impaired Cas9 nickase (nCas9, typically D10A) tethered to an engineered adenine deaminase enzyme (e.g., TadA-8e). The nCas9 moiety programmably targets the complex to a specific genomic locus via a single-guide RNA (sgRNA). Within the transient, single-stranded DNA R-loop formed upon binding, the deaminase domain converts an adenine (A) within a defined window (typically positions 4-8, counting the PAM as 21-23) to inosine (I). Cellular DNA repair machinery interprets inosine as guanine (G), leading to the permanent A•T to G•C change upon replication. The complementary strand is nicked by the nCas9 to bias repair toward the edited strand.
Diagram 1: ABE Mechanism and Cellular Repair
Quantitative Landscape of Targetable Diseases
A significant proportion of human genetic diseases are caused by point mutations amenable to correction by ABEs. The table below summarizes key disease targets, their associated genes, and the specific A•T to G•C corrections required.
Table 1: Exemplary Genetic Diseases Targetable by ABE Correction
| Disease | Gene | Pathogenic Mutation (Genomic) | Required Correction (A•T to G•C) | Reference (Sample) |
|---|---|---|---|---|
| Hereditary Hemochromatosis | HFE | c.845G>A (p.Cys282Tyr) | Corrects the C allele (G) to T (A on reverse strand)* | Gaudelli et al., 2017 |
| Hutchinson-Gilford Progeria | LMNA | c.1824C>T (p.Gly608Gly) | Corrects T (A) to C (G) in exon 11 | Koblan et al., 2021 |
| Sickle Cell Disease / β-Thalassemia | HBB | c.-78A>G (promoter) | Corrects G (C) to A (T on reverse strand)* to upregulate HbF | Newby et al., 2021 |
| Niemann-Pick Disease Type C | NPC1 | c.3182T>C (p.Ile1061Thr) | Corrects C (G) to T (A on reverse strand)* | Grünewald et al., 2020 |
| Duchenne Muscular Dystrophy | DMD | c.10009G>A (p.Glu3337Lys) | Corrects A (T) to G (C) for exon-skipping | Ryu et al., 2018 |
| Deafness | TMC1 | c.1234A>G (p.Thr412Ala) | Corrects G (C) to A (T on reverse strand)* in Beethoven model | Gao et al., 2018 |
Note: ABEs act on the non-complementary strand within the R-loop. The indicated "correction" is the genomic outcome.
Core Experimental Protocol forIn VitroABE Editing
This protocol outlines key steps for evaluating ABE efficacy in cultured cells.
A. Design and Cloning
- sgRNA Design: Design a 20-nt spacer sequence targeting the nCas9 (D10A) to the genomic locus of interest, ensuring the target adenine (A) is within positions 4-8 (protospacer position, PAM-distal end as position 1).
- Cloning: Clone the sgRNA sequence into an appropriate expression plasmid (e.g., pCMV_ABE8e, Addgene #138489) using BsaI Golden Gate assembly or ligation.
B. Cell Transfection and Culture
- Cell Seeding: Seed HEK293T or other relevant cell line (e.g., patient-derived fibroblasts) in a 24-well plate to reach 70-80% confluence at transfection.
- Transfection Complex Formation: For each well, combine 500 ng of ABE expression plasmid and 250 ng of sgRNA plasmid (or a single all-in-one plasmid) with 1.5 µL of polyethylenimine (PEI, 1 µg/µL) in 50 µL of Opti-MEM. Incubate for 15 min.
- Transfection: Add the complex dropwise to cells in complete medium.
- Harvest: Incubate cells for 72-96 hours to allow for editing and protein turnover. Harvest cells by trypsinization.
C. Analysis of Editing Outcomes
- Genomic DNA Extraction: Use a silica-column-based kit to extract gDNA.
- PCR Amplification: Amplify the target locus using high-fidelity polymerase. Primer design should yield an ~500-800 bp product.
- Sequencing and Analysis:
- Sanger Sequencing: Purify PCR product and sequence. Analyze chromatograms for trace overlays at the target base using decomposition software (e.g., EditR or BE-Analyzer).
- Next-Generation Sequencing (NGS): Index PCR amplicons and perform paired-end sequencing (MiSeq). Align reads to the reference genome and quantify the percentage of A-to-G conversion using pipelines like CRISPResso2.
Diagram 2: ABE Experimental Workflow
The Scientist's Toolkit: Key Reagent Solutions
Table 2: Essential Research Reagents for ABE Experiments
| Reagent / Material | Function & Description | Example Product/Catalog |
|---|---|---|
| ABE Expression Plasmid | Encodes the nCas9 (D10A)-TadA fusion protein under a constitutive/inducible promoter. | pCMV_ABE8e (Addgene #138489) |
| sgRNA Cloning Backbone | Plasmid for expressing sgRNA under a U6 promoter. | pGL3-U6-sgRNA (Addgene #51133) |
| All-in-one ABE Vector | Combines ABE and sgRNA expression in a single plasmid for simplified delivery. | pAAV-ABE8e (various) |
| Delivery Reagent | For plasmid transfection in vitro (e.g., lipofection, electroporation reagents). | Lipofectamine 3000, PEI Max, Neon Electroporation System |
| High-Fidelity Polymerase | For error-free amplification of the target locus prior to sequencing. | Q5 Hot-Start (NEB), KAPA HiFi |
| NGS Library Prep Kit | For preparing amplicon libraries to quantify editing efficiency and byproducts. | Illumina DNA Prep |
| EditR / BE-Analyzer | Software tools for analyzing Sanger sequencing traces to quantify base editing. | (Web-based tools) |
| CRISPResso2 | Bioinformatics pipeline for quantifying editing outcomes from NGS data. | (Python package) |
| Control gDNA | Unedited wild-type and heterozygous/homozygous mutant genomic DNA for assay calibration. | Coriell Institute Biorepository |
Advanced Considerations and Challenges
- Off-Target Editing: ABEs can cause off-target edits in both DNA and RNA. Employ techniques like GUIDE-seq or Digenome-seq for DNA off-target profiling, and use engineered high-fidelity Cas9 domains (e.g., HiFi Cas9) or evolved TadA variants with improved specificity.
- Product Purity: Undesired byproducts include low levels of indel formation from the nickase activity and non-A-to-G edits (e.g., A-to-C, A-to-T). Optimization of editor version (e.g., ABE8e vs. ABE7.10), delivery method, and expression levels is critical.
- In Vivo Delivery: Therapeutic translation requires efficient, safe delivery vehicles. Adeno-associated virus (AAV) is a common vector but has a limited packaging capacity (~4.7 kb), necessitating the use of split-intein systems or more compact editors (e.g., SaABE). Lipid nanoparticles (LNPs) are an emerging non-viral alternative.
This technical guide details methodologies for creating saturation mutagenesis libraries and applying them to protein engineering, framed within the core thesis inquiry: How do adenine base editors (ABEs) work? ABEs, which catalyze the direct, irreversible conversion of A•T to G•C base pairs without causing double-stranded DNA breaks, serve as both a premier application and a driving force for these functional genomics techniques.
Core Principles: ABE Mechanism as a Case Study
Adenine Base Editors (ABEs) are fusion proteins combining a catalytically impaired CRISPR-Cas9 nickase (nCas9) with an engineered adenine deaminase enzyme (e.g., TadA-8e). The editor localizes to a target DNA sequence via a guide RNA (gRNA). Within a narrow window (typically positions 4-8, counting the PAM as 21-23), the deaminase converts adenine (A) to inosine (I). Cellular machinery interprets inosine as guanosine (G), resulting in a permanent A•T to G•C change upon DNA replication or repair.
Diagram 1: ABE Mechanism and Outcome
Creating Saturation Mutagenesis Libraries with Base Editors
Saturation mutagenesis aims to create all possible single-nucleotide variants at a defined genomic locus or protein coding sequence. ABEs enable precise, efficient C•G to T•A (using CBEs) and A•T to G•C (using ABEs) saturation.
Table 1: Comparison of Saturation Mutagenesis Library Generation Methods
| Method | Core Principle | Compatible with ABEs? | Key Advantage | Primary Limitation |
|---|---|---|---|---|
| Oligo Pool Synthesis | Synthetic oligonucleotides encoding all variants are cloned en masse. | Indirectly (as template) | Comprehensive, user-defined control over variants. | High cost for large genes; cloning bottleneck. |
| Error-Prone PCR | PCR with low-fidelity polymerases introduces random mutations. | No | Simple, fast, no prior sequence knowledge needed. | Biased mutation spectrum (A/T > G/C); not truly saturating. |
| CRISPR-Cas9 HDR | Cas9 cleavage followed by homology-directed repair with oligo donors. | Can be combined | Precise insertion of any mutation, including non-natural AAs. | Low efficiency, requires dsDNA cleavage. |
| Base Editor gRNA Libraries | Delivering a library of gRNAs tiling across a target region with an ABE. | Yes | Highly efficient, simultaneous generation of all possible A>G or T>C changes. | Restricted to canonical BE outcomes (A>G, C>T). |
Protocol 2.1: Saturation Mutagenesis Using an ABE gRNA Library Objective: Generate all possible A>G (sense strand) mutations within a target protein domain.
- Target Region Analysis: Define the amino acid codons for mutagenesis. Identify all adenine (A) bases on the sense strand within the editing window (e.g., protospacer positions 4-8, relative to the NGG PAM) for each possible gRNA.
- gRNA Library Design: Synthesize a pooled oligonucleotide library where each oligo encodes a unique gRNA spacer sequence targeting an individual A residue. Include constant regions for cloning into your chosen gRNA expression backbone (e.g., U6 promoter).
- Library Cloning: Use a highly efficient Golden Gate or Gibson assembly to clone the pooled oligo library into the expression vector. Transform into a competent E. coli strain optimized for library diversity (e.g., EndA- cells). Harvest plasmid DNA from >10x library size colonies to ensure representation.
- Delivery & Editing: Co-transfect the pooled gRNA library plasmid with the ABE expression plasmid (e.g., ABE8e) into the target cell line at a multiplicity ensuring single-copy delivery (e.g., low MOI for lentiviral delivery).
- Harvest & Sequence: After 3-7 days, extract genomic DNA. Amplify the target region via PCR and subject to next-generation sequencing (NGS). Analyze the frequency of A>G transitions at each targeted position.
Protein Engineering via Base Editor-Mediated Functional Screens
ABE-generated saturation libraries can be coupled with phenotypic screens to identify protein variants with enhanced or novel functions.
Diagram 2: ABE Library Screen for Protein Engineering
Protocol 3.1: Enrichment Screen for Drug-Resistant Protein Variants Objective: Identify mutations in a target protein that confer resistance to an inhibitory drug.
- Library Creation & Editing: Create and deliver an ABE saturation mutagenesis library for your target gene as in Protocol 2.1.
- Selection: At 7 days post-editing (allowing for protein turnover), split cells into two groups: a control group and a group treated with the inhibitory drug at a lethal concentration (e.g., IC90). Maintain drug selection for 10-14 days, replenishing as needed.
- Genomic DNA Extraction: Harvest genomic DNA from the pre-selection library (reference), the drug-treated population, and the untreated control population.
- Amplicon Sequencing & Analysis: Amplify the target region and perform deep sequencing (>500x coverage). Calculate the frequency of each A>G mutation in each sample. Identify mutations significantly enriched in the drug-treated population compared to both the pre-selection and untreated controls using statistical models (e.g., Fisher's exact test, MAGeCK).
Table 2: Key Quantitative Metrics from a Representative ABE Saturation Screen
| Metric | Typical Value / Result | Interpretation |
|---|---|---|
| Library Coverage | >1000x per gRNA | Ensures statistical power to detect variants. |
| Average Editing Efficiency | 20-50% per target A (for ABE8e) | Fraction of alleles successfully modified. |
| Background Mutation Rate | <0.1% at non-targeted bases | Confirms specificity of ABE activity. |
| Fold-Enrichment (Hit Threshold) | >10-fold, p < 0.001 | Identifies high-confidence functional variants. |
| Primary Hit Rate | 0.1% - 5% of tested sites | Proportion of mutated residues affecting function under selection. |
The Scientist's Toolkit: Essential Reagents for ABE Saturation Libraries
Table 3: Research Reagent Solutions for ABE-Mediated Saturation Mutagenesis
| Item | Function in the Experiment |
|---|---|
| High-Fidelity DNA Polymerase (e.g., Q5, KAPA HiFi) | Accurate amplification of target loci for NGS library prep and cloning steps. |
| Pooled gRNA Library Oligos | Commercially synthesized oligo pool encoding all designed spacer sequences. |
| gRNA Expression Backbone (e.g., lentiviral U6 vector) | Plasmid for high-efficiency cloning and expression of the gRNA library. |
| ABE Expression Plasmid (e.g., ABE8e-NLS) | Source of the adenine base editor protein (nCas9-TadA fusion). |
| Ultracompetent E. coli (EndA-) | For high-efficiency transformation to maintain library complexity during plasmid prep. |
| Lentiviral Packaging System (psPAX2, pMD2.G) | For generating viral particles to deliver the gRNA library and ABE stably/in vitro. |
| Next-Generation Sequencing Kit (e.g., Illumina) | For preparing and sequencing amplicons from edited cell populations. |
| Cell Selection Agent (e.g., Puromycin, Drug Inhibitor) | For stable cell line generation and phenotypic screening pressure. |
| Genomic DNA Extraction Kit | High-yield, pure gDNA preparation from edited cell pools for downstream NGS. |
| CRISPR Enrichment Analysis Software (e.g., MAGeCK, CRISPResso2) | Bioinformatics tools to calculate editing efficiencies and identify enriched hits from NGS data. |
Saturation mutagenesis libraries, powered by the precision and efficiency of adenine base editors, have revolutionized protein engineering. By systematically converting A•T to G•C base pairs, ABEs enable the direct exploration of protein sequence-function relationships at single-amino-acid resolution. The integration of ABE-generated variant libraries with robust phenotypic screens provides a powerful, scalable pipeline for discovering protein variants with optimized properties, directly addressing the mechanistic question of how specific residues contribute to function and offering invaluable insights for therapeutic development.
Optimizing ABE Experiments: Solving Common Problems with Efficiency, Specificity, and Delivery
Diagnosing and Improving Low Base Editing Efficiency
Base editing is a powerful CRISPR-derived technology that enables the direct, irreversible conversion of one DNA base pair to another without requiring double-stranded DNA breaks (DSBs) or donor DNA templates. Within this field, Adenine Base Editors (ABEs) catalyze the conversion of an A•T base pair to a G•C base pair. This guide, framed within the context of understanding How do adenine base editors (ABEs) work?, provides a systematic, technical approach for diagnosing and remediating low editing efficiency in experimental settings.
Core Mechanism of Adenine Base Editors (ABEs)
ABEs are fusion proteins consisting of a catalytically impaired Cas9 nickase (nCas9) tethered to an engineered adenine deaminase enzyme. The nCas9 component is guided by a single guide RNA (sgRNA) to a target genomic locus, where it unwinds the DNA duplex, exposing a single-stranded DNA (ssDNA) region within the R-loop. The adenine deaminase acts on this exposed ssDNA to convert adenine (A) to inosine (I). Cellular DNA repair machinery interprets inosine as guanosine (G), leading to the permanent replacement of the A•T base pair with a G•C base pair. The canonical editing window is typically positions 4-8 within the protospacer, relative to the protospacer adjacent motif (PAM).
Diagram: ABE Catalytic Mechanism
Diagnostic Framework for Low Efficiency
Low base editing efficiency can stem from multiple factors. The following table outlines primary diagnostic categories, potential causes, and corresponding validation assays.
Table 1: Diagnostic Framework for Low ABE Efficiency
| Category | Potential Cause | Validation Assay |
|---|---|---|
| Editor Expression & Delivery | Low transfection/transduction efficiency; Poor nuclear localization; Protein instability. | Western blot for ABE protein; Flow cytometry for fluorescent reporter (if fused). |
| sgRNA Design & Activity | Suboptimal sgRNA sequence; Low transcription/expression; Incorrect targeting. | NGS of target locus (check indels from nicking); qRT-PCR for sgRNA levels. |
| Target Sequence Context | Epigenetic silencing (e.g., heterochromatin); Local DNA secondary structure; Unfavorable sequence for deaminase. | ATAC-seq/ChIP for chromatin accessibility; In vitro deamination assay. |
| Cellular Context | Low activity of repair pathways; Cell cycle phase; High endogenous deaminase inhibitor expression. | Cell cycle analysis; RNA-seq for repair gene expression. |
| Experimental Design | Inadequate delivery time; Suboptimal dosage; Inefficient readout method. | Time-course experiment; Dose-response curve. |
Key Experimental Protocols for Diagnosis
Protocol 3.1: Quantifying ABE Protein Expression (Western Blot)
- Harvest Cells: 48-72h post-transfection/transduction.
- Lysis: Use RIPA buffer with protease inhibitors.
- Gel Electrophoresis: Load 20-40 µg protein on a 4-12% Bis-Tris gel.
- Transfer: Transfer to PVDF membrane.
- Blotting: Probe with primary antibody against the deaminase domain (e.g., TadA) or epitope tag (e.g., HA, FLAG). Use anti-Cas9 antibody with caution due to size.
- Normalization: Re-probe for a housekeeping protein (e.g., GAPDH, β-Actin).
Protocol 3.2: Assessing On-Target Activity & Byproduct Formation (Next-Generation Sequencing)
- PCR Amplification: Design primers flanking the target site (amplicon size: 200-300 bp).
- Library Preparation: Use a two-step PCR protocol with addition of sample barcodes and sequencing adapters.
- Sequencing: Perform paired-end sequencing on an Illumina MiSeq or equivalent.
- Analysis: Use pipelines like CRISPResso2 or BE-Analyzer to quantify:
- Percentage of A-to-G conversions within the editing window.
- Percentage of indels (from Cas9 nickase activity).
- Other nucleotide substitutions (potential off-target deamination).
Table 2: Example NGS Analysis Output for ABE Evaluation
| Sample | Total Reads | % A-to-G (Edits) | % Indels | % Other Subs | Interpretation |
|---|---|---|---|---|---|
| ABE_v1 + sgRNA1 | 50,000 | 8.5 | 1.2 | 0.3 | Low efficiency. |
| ABE_v1 + sgRNA2 | 48,000 | 45.7 | 1.5 | 0.4 | Good efficiency. |
| ABE_v2 + sgRNA1 | 52,000 | 22.3 | 0.8 | 0.2 | Improved efficiency. |
| Untreated Control | 49,000 | 0.1 | 0.05 | 0.1 | Background. |
Protocol 3.3:In VitroDeamination Assay (Assessing Enzyme Activity)
- Prepare Substrate: Synthesize a fluorescently-labeled ssDNA oligonucleotide containing the target adenine(s).
- Assemble Reaction: Incubate purified ABE protein (or cell lysate) with substrate oligonucleotide in reaction buffer.
- Stop Reaction: Add UDG (uracil DNA glycosylase) to cleave any deoxyinosine bases, followed by NaOH to fragment the backbone at abasic sites.
- Analyze: Run products on a denaturing PAGE gel. Cleavage products indicate deamination activity.
Diagram: Diagnostic Workflow for Low ABE Efficiency
Strategies for Improving ABE Efficiency
Based on diagnostic results, implement targeted improvements.
Table 3: Optimization Strategies and Their Applications
| Strategy | Method | Expected Outcome | Considerations |
|---|---|---|---|
| Optimize Delivery | Use electroporation for primary cells; optimize viral titer (MOI); use protein-RNP delivery. | Increased percentage of editor-positive cells. | RNP delivery reduces persistence, useful for reducing off-targets. |
| Enhance sgRNA Activity | Use modified sgRNAs (e.g., MS2, eRNA); optimize Pol III promoter (U6, H1); test truncated sgRNAs. | Higher nuclear sgRNA concentration, improved binding. | Chemical modifications can enhance stability. |
| Modulate Chromatin | Co-express chromatin-opening factors (e.g., VP64, p300); use histone deacetylase inhibitors (e.g., VPA). | Increased accessibility of target locus. | Risk of global transcriptional changes. |
| Utilize Improved ABE Variants | Use next-gen editors (e.g., ABE8e, ABE9e) with higher activity or altered editing windows. | Significantly higher efficiency on stubborn targets. | Potential for increased RNA or sequence-context off-targets. |
| Leverage Cell Cycle | Synchronize cells or use cell cycle regulators; S-phase often shows higher efficiency. | Improved engagement of DNA repair machinery. | Impractical for some cell types. |
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Reagents for ABE Experiments
| Reagent/Material | Function/Description | Example Vendor/Product |
|---|---|---|
| ABE Plasmid or mRNA | Expresses the adenine base editor protein. Can be all-in-one (editor + sgRNA) or separate. | Addgene (e.g., pCMV_ABE8e); TriLink BioTechnologies (CleanCap mRNA). |
| sgRNA Expression Vector | Drives expression of the single guide RNA under a U6 or H1 promoter. | Synthego (custom sgRNA); Integrated DNA Technologies (gBlocks). |
| Delivery Reagent | Facilitates intracellular delivery of nucleic acids (plasmid, mRNA, RNP). | Lonza (Nucleofector kits); Thermo Fisher (Lipofectamine CRISPRMAX). |
| Purified ABE Protein | For ribonucleoprotein (RNP) complex delivery. Rapid action, reduced off-target persistence. | Aldevron; Thermo Fisher (TrueCut Cas9 Protein). |
| NGS Library Prep Kit | For preparation of amplicon sequencing libraries to quantify editing. | Illumina (Nextera XT); New England Biolabs (NEBNext Ultra II). |
| Anti-Cas9/Anti-Tag Antibody | For detecting ABE protein expression via Western blot or immunofluorescence. | Cell Signaling Technology (Anti-Cas9 #14697); Sigma (Anti-FLAG M2). |
| Chromatin Modulator | Small molecule to test the effect of chromatin state on editing. | Cayman Chemical (Valproic Acid); Tocris (Trichostatin A). |
| Control Plasmid (Fluorescent) | To monitor and normalize for transfection/transduction efficiency. | Addgene (e.g., pMAX-GFP). |
Diagram: Optimization Strategy Decision Tree
Adenine Base Editors (ABEs) represent a transformative advancement in precision genome editing, enabling the direct, irreversible conversion of A•T base pairs to G•C without requiring double-stranded DNA breaks. The core mechanism involves a catalytically impaired Cas9 nickase (Cas9n) fused to an engineered adenine deaminase enzyme (TadA). This complex binds to a target DNA sequence specified by a single-guide RNA (sgRNA), where the deaminase domain catalyzes the conversion of adenosine (A) to inosine (I) within a defined activity window (typically positions 4-8 within the protospacer). Inosine is subsequently read as guanosine (G) by cellular machinery, completing the edit.
Within the broader thesis on "How do adenine base editors (ABEs) work?", a critical pillar of investigation is the characterization and minimization of off-target effects. While the nCas9 component reduces off-target DSBs compared to wild-type Cas9, ABEs can still catalyze off-target edits at both the DNA and RNA levels. DNA off-targets occur at genomic loci with sequence homology to the sgRNA. RNA off-targets result from promiscuous deamination of adenosines in cellular RNA by the TadA domain. Comprehensive analysis strategies for both are essential for evaluating the safety and specificity of ABE tools for research and therapeutic applications.
DNA Off-Target Analysis Strategies
DNA off-target editing arises from the Cas9-sgRNA complex binding to genomic sites with imperfect complementarity. Analysis focuses on prediction, detection, and quantification.
PredictiveIn SilicoAnalysis
Computational tools predict potential off-target sites based on sequence similarity to the on-target sgRNA.
- Common Tools: Cas-OFFinder, COSMID, CRISPRseek.
- Method: Input the sgRNA sequence and specify the number of mismatches, bulges, and the PAM (NGG for SpCas9). The tool scans the reference genome to return a list of putative off-target sites.
- Limitation: Relies on a reference genome and may miss sites with structural variants or in non-canonical PAM contexts exploited by some Cas9 variants.
Empirical, Genome-Wide Detection Methods
These unbiased methods experimentally identify off-target sites across the genome.
a) CIRCLE-Seq (Circularization for In vitro Reporting of Cleavage Effects by Sequencing) Adapted for base editors by using nCas9 and sequencing to detect binding/accessibility rather than cleavage.
Protocol:
- Genomic DNA Isolation: Extract high-molecular-weight genomic DNA from cells or tissue of interest.
- In vitro RNP Complex Formation: Form ribonucleoprotein (RNP) complexes with purified ABE protein and the target sgRNA.
- In vitro Editing Reaction: Incubate RNP complexes with the sheared genomic DNA under optimal reaction conditions (buffer, temperature, time).
- DNA Circularization: Thoroughly purify DNA to remove proteins. Use exonuclease to degrade linear DNA, enriching circular DNA fragments that were bound/edited by ABE.
- Library Preparation & Sequencing: Linearize circular DNA, add sequencing adapters, and perform high-throughput sequencing.
- Data Analysis: Align sequences to the reference genome and identify sites with A-to-G conversions above background, excluding the intended on-target site.
b) GUIDE-Seq (Genome-wide, Unbiased Identification of DSBs Enabled by Sequencing) Originally for nucleases, it can be adapted for base editors by co-delivering a nuclease to create DSBs at ABE-bound off-target sites, though this is indirect.
c) Digenome-Seq Adapted for base editors by using nCas9 and whole-genome sequencing (WGS) of *in vitro treated genomic DNA.*
Protocol:
- In vitro Treatment: Incubate purified genomic DNA with ABE RNP.
- Whole-Genome Sequencing: Subject the treated DNA and an untreated control to high-coverage WGS (>80x).
- Variant Calling: Use a sensitive variant caller (e.g., GATK) to identify all A-to-G substitutions.
- Off-Target Identification: Filter variants present only in the treated sample, subtract known SNPs (using dbSNP), and filter out the on-target site. Remaining sites are candidate off-targets, which must be validated in cellula.
Table 1: Comparison of Genome-Wide DNA Off-Target Detection Methods
| Method | Principle | Key Advantage | Key Limitation | Typical Readout |
|---|---|---|---|---|
| CIRCLE-Seq | In vitro editing & circularization | High sensitivity; low background | Purely in vitro; may not reflect chromatin state | List of off-target sites with read counts |
| GUIDE-Seq | Integration of double-stranded oligos at DSBs in cells | Captures cellular context (chromatin, etc.) | Requires co-transfection of nuclease; indirect for ABE | List of off-target sites with tag integration counts |
| Digenome-Seq | Whole-genome sequencing after in vitro treatment | Truly genome-wide; no sequence bias | Expensive (high-coverage WGS); high computational load | List of genomic coordinates with A-to-G variant frequency |
| SITE-Seq | In vitro cleavage/blunting/ligation of nCas9-bound sites | Biochemical mapping of binding events | Does not directly measure editing outcome | List of off-target binding sites |
RNA Off-Target Analysis Strategies
The TadA deaminase domain, evolved from E. coli TadA, can deaminate adenosines in single-stranded RNA, leading to transcriptome-wide A-to-I (read as G) edits.
Primary Method: RNA-Sequencing (RNA-Seq) Protocol for ABE RNA Off-Target Analysis:
- Cell Transfection/Transduction: Deliver ABE and target sgRNA (or non-targeting control) into relevant cell lines. Include a control expressing nCas9 without TadA.
- RNA Harvest: 48-72 hours post-delivery, extract total RNA using a column-based method with DNase I treatment.
- RNA-Seq Library Prep: Use a strand-specific library preparation kit. Ribo-depletion is preferred over poly-A selection to capture non-coding RNAs.
- High-Throughput Sequencing: Perform paired-end sequencing (≥50 million reads per sample) to sufficient depth.
- Bioinformatic Analysis:
- Alignment: Map reads to the reference transcriptome using a splice-aware aligner (e.g., STAR).
- Variant Calling: Use specialized tools (e.g., GATK's ASEReadCounter, REDItools) to call A-to-G mismatches from the aligned BAM files.
- Filtering: Rigorously filter artifacts by:
- Subtracting variants found in the nCas9-only control.
- Removing known RNA editing sites (from databases like REDIportal).
- Applying a minimum read depth (e.g., ≥10) and variant frequency threshold (e.g., ≥0.1%).
- Focusing on sites with a statistically significant increase in A-to-G edits in ABE samples vs. all controls.
Table 2: Quantifying RNA Off-Target Effects of ABE Variants
| ABE Variant (Example) | Median Transcriptome-Wide A-to-I Editing % (from RNA-seq) | Key Differentiating Feature | Reference (Recent) |
|---|---|---|---|
| ABE7.10 | ~0.03 - 0.15% | First-generation, high activity | Rees et al., 2019 |
| ABE8e | < 0.02% | Engineered TadA dimer; reduced RNA off-targets | Richter et al., 2020 |
| ABE8s | Undetectable (near background) | Single TadA construct; superior RNA specificity | Gaudelli et al., 2020 / Richter et al., 2020 |
Integrated Off-Target Profiling and Mitigation
Best practice involves a combinatorial approach: using in silico prediction to guide focused screening, followed by genome-wide empirical methods for DNA, and transcriptome sequencing for RNA. The ultimate goal is mitigation.
Mitigation Strategies:
- Optimized sgRNA Design: Choose sgRNAs with minimal homology to other genomic sites using predictive algorithms.
- Engineered Editor Variants: Use next-generation ABEs with demonstrated lower off-target profiles (e.g., ABE8e, ABE8s).
- Delivery Method & Dose: Use RNP delivery or limit expression time (e.g., with mRNA) to reduce off-target editing, which is often dose- and time-dependent.
- Contextual Evaluation: Always profile off-targets in therapeutically relevant cell types, as chromatin accessibility varies.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for ABE Off-Target Analysis
| Item/Category | Function in Off-Target Analysis | Example Product/Note |
|---|---|---|
| High-Fidelity ABE Plasmids/mRNAs | Expressing the editor with minimal sequence errors. | Addgene: ABE8e (plasmid #138495); TriLink CleanCap ABE mRNA. |
| Control sgRNAs | Critical for distinguishing background. | Non-targeting control sgRNA (scrambled sequence). |
| Next-Generation ABE Variants | Editors with inherently lower off-target risk. | ABE8s, ABE9e for reduced RNA/DNA off-targets. |
| In vitro Transcribed sgRNA Kit | For forming RNP complexes in CIRCLE-Seq/Digenome-Seq. | NEB HiScribe T7 Quick High Yield Kit. |
| Purified nCas9-ABE Protein | For in vitro off-target assays (CIRCLE-Seq). | Commercial SpCas9-ABE protein (e.g., from ToolGen). |
| Genomic DNA Isolation Kit (for cells) | High-quality, high-molecular-weight DNA for in vitro assays. | Qiagen Blood & Cell Culture DNA Maxi Kit. |
| CIRCLE-Seq Kit | Optimized reagents for the CIRCLE-Seq protocol. | Varies; often performed with core lab reagents. |
| Whole-Genome Sequencing Service | For Digenome-Seq; requires ultra-deep coverage. | Illumina NovaSeq, PacBio HiFi for complex regions. |
| Ribo-depletion RNA-Seq Kit | For transcriptome-wide RNA off-target detection. | Illumina Stranded Total RNA Prep with Ribo-Zero Plus. |
| Targeted Amplicon-Seq Kit | Orthogonal validation of candidate off-target sites. | Illumina DNA Prep with custom amplification primers. |
| NGS Analysis Software | For variant calling and filtering. | GATK, CRISPResso2, REDItools (for RNA). |
Adenine Base Editors (ABEs) enable the direct, programmable conversion of A•T to G•C base pairs without introducing double-strand DNA breaks (DSBs). While offering a transformative tool for research and therapeutic development, ABE applications are complicated by notable byproducts: insertion-deletion mutations (indels) and undesired base conversions (e.g., A-to-G efficiency, A-to-C/G transversions). This technical guide details the mechanistic origins of these byproducts and provides validated, current experimental strategies for their minimization, thereby enhancing editing purity.
ABEs are fusion proteins consisting of a catalytically impaired Cas9 nickase (nCas9) and an engineered adenosine deaminase enzyme (e.g., TadA-8e). The complex binds to a target DNA sequence specified by a single-guide RNA (sgRNA), locally unwinding the DNA to form an R-loop. The deaminase domain then converts adenosine (A) to inosine (I) within a defined activity window (typically positions 4-8, counting the PAM as 21-23). Inosine is read as guanosine (G) by cellular polymerases and repair machinery, resulting in an A•T to G•C change. However, two primary byproducts arise:
- Indels: Resulting from the nCas9-induced nick on the non-edited strand, which can be misinterpreted by cellular repair pathways as a DSB intermediate.
- Undesired Base Conversions: Including low-efficiency A-to-G editing and rarer A-to-C or A-to-T transversions, often stemming from off-target deamination or error-prone repair.
Quantitative Analysis of Byproduct Frequencies
Recent studies quantify the baseline rates of these byproducts under standard ABE editing conditions (e.g., ABE8e with a canonical sgRNA).
Table 1: Typical Byproduct Frequencies for ABE8e in Mammalian Cells
| Byproduct Type | Typical Frequency Range | Primary Cause |
|---|---|---|
| Desired A-to-G Conversion | 20-60% (highly target-dependent) | TadA-mediated deamination. |
| Indels | 0.1 - 3.0% | Nick-induced mismatch repair or BER processing. |
| A-to-C / A-to-T Transversions | <0.1 - 0.5% | Potentially from inosine mispairing or polymerase errors. |
| Other Base Edits (C, T) | <0.1% | Off-target deamination or background noise. |
Experimental Protocols for Byproduct Assessment
Accurate quantification is essential for evaluating mitigation strategies.
Protocol 1: Amplicon Sequencing for Byproduct Analysis
- Design Primers: Design PCR primers (with overhangs for indexing) to amplify a ~300-500 bp region surrounding the target site.
- Extract Genomic DNA: 72 hours post-transfection, harvest cells and extract high-quality gDNA.
- PCR Amplification: Perform two-step PCR. First, amplify target locus. Second, add dual-indexing barcodes and sequencing adapters.
- Library Purification & Quantification: Use magnetic beads for size selection and quantify via qPCR.
- High-Throughput Sequencing: Sequence on an Illumina MiSeq or NovaSeq platform (≥ 10,000x depth per sample).
- Data Analysis: Use pipelines like CRISPResso2 or BE-Analyzer to quantify base substitution frequencies and indel percentages at the target locus.
Protocol 2: In Vitro Deamination Assay for Specificity Profiling
- Purify ABE Protein: Express and purify His-tagged ABE protein from E. coli or HEK293 cells.
- Prepare dsDNA Substrate: Generate a fluorescently labeled dsDNA oligo containing the target sequence.
- Reaction Setup: Incubate ABE protein (100 nM) with dsDNA substrate (50 nM) in reaction buffer (20 mM HEPES, 150 mM KCl, 1 mM DTT, pH 7.5) at 37°C for 1 hour.
- Reaction Stop & Digestion: Add proteinase K to stop, then digest with UDG and Endonuclease VIII to cleave at inosine sites.
- Fragment Analysis: Run products on capillary electrophoresis (e.g., ABI 3730xl). Cleavage fragment sizes reveal deamination positions and frequency, providing a profile without cellular repair confounders.
Strategies for Minimizing Byproducts
4.1. Reducing Indel Formation
- Rationale: Indels arise primarily from prolonged exposure of the nCas9-induced nick.
- Strategy 1: Use of DNA Ligase Inhibitors. Transient treatment with Scr7 (a DNA Ligase IV inhibitor) can bias repair away from NHEJ-like pathways at the nick site.
- Protocol: Add 1µM Scr7 to culture media 24 hours post-transfection; maintain for 48 hours before analysis.
- Strategy 2: Engineered Cas Variants. Use Cas9 mutants with reduced nicking activity on the non-edited strand or altered conformational states that reduce nick exposure time.
4.2. Minimizing Undesired Base Conversions
- Rationale: Transversions may result from error-prone translation of inosine or deamination of non-adenine bases.
- Strategy 1: Optimized ABE Architecture. Fuse the deaminase domain via longer, more flexible linkers to prevent steric strain that may promote off-target deamination within the R-loop.
- Strategy 2: High-Fidelity TadA Variants. Employ computationally re-designed TadA variants (e.g., ABE8.8, ABE8.17) with increased specificity for adenosine over cytosine.
- Strategy 3: sgRNA Engineering. Modify sgRNA length (e.g., using 18-19 nt spacers instead of 20 nt) to alter R-loop dynamics and narrow the deamination activity window.
Visualizing Pathways and Workflows
Title: ABE Editing and Byproduct Generation Pathway
Title: Workflow for Assessing ABE Byproducts
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents for ABE Byproduct Studies
| Reagent/Material | Function & Importance | Example Product/Catalog |
|---|---|---|
| High-Fidelity ABE Plasmids | Express engineered ABE variants (e.g., ABE8.8) with improved specificity. | pCMV_ABE8.8 (Addgene #138495) |
| Chemically Modified sgRNA | Enhances stability and editing efficiency; truncated variants can narrow editing window. | Synthetic sgRNA with 2'-O-methyl modifications. |
| Next-Generation Sequencing Kit | For precise, quantitative measurement of editing outcomes and byproducts. | Illumina DNA Prep Kit |
| CRISPResso2 Software | Critical open-source tool for analyzing deep sequencing data of base editing outcomes. | Available on GitHub |
| DNA Ligase IV Inhibitor (Scr7) | Small molecule to transiently inhibit error-prone end-joining at nick sites, reducing indels. | SCR7 (HY-13289, MedChemExpress) |
| Recombinant ABE Protein | For in vitro kinetics and specificity assays without cellular repair variables. | Custom purification from HEK293 or E. coli. |
| UDG & Endonuclease VIII Mix | Enzymes for cleaving at inosine sites in in vitro deamination assays. | USER Enzyme (NEB M5505) |
Optimizing Delivery Efficiency for Challenging Cell Types and In Vivo Use
The development of Adenine Base Editors (ABEs) represents a transformative advancement in precision genome editing, enabling the direct, irreversible conversion of A•T to G•C base pairs without inducing double-strand DNA breaks. The core thesis of ABE research investigates the molecular architecture and mechanism of these fusion proteins, which typically consist of a catalytically impaired CRISPR-Cas nuclease (e.g., dCas9 or a nickase) tethered to an engineered adenine deaminase enzyme (e.g., TadA). While the efficiency and fidelity of the editor itself are paramount, the broader applicability of this technology—especially for therapeutic interventions—is critically dependent on the delivery vehicle. This guide focuses on the pivotal challenge of delivering ABE ribonucleoprotein (RNP) complexes or nucleic acid templates into challenging primary cells (e.g., hematopoietic stem cells, neurons, T cells) and for in vivo applications, where delivery efficiency and specificity are the primary bottlenecks.
Quantitative Comparison of Delivery Modalities
The choice of delivery method is dictated by target cell type, required editing efficiency, off-target risk, and immunogenicity. The table below summarizes key performance metrics for current leading platforms.
Table 1: Delivery Platform Performance for ABEs
| Delivery Modality | Typical Format for ABEs | Max In Vitro Efficiency (Primary Cells) | Key In Vivo Route | Immunogenicity Concern | Payload Capacity | Key Challenge Addressed |
|---|---|---|---|---|---|---|
| Viral Vectors (AAV) | DNA (plasmid or split-inteins) | 20-60% (hepatocytes) | Systemic, local | High (capsid, cargo) | Low (<~4.7 kb) | Long-term expression, in vivo targeting |
| Electroporation (Nucleofection) | RNP or mRNA/sgRNA | 40-90% (immune cells, HSCs) | Ex vivo only | Low | High | Hard-to-transfect primary cells |
| Lipid Nanoparticles (LNPs) | mRNA/sgRNA or RNP | 30-70% (hepatocytes) | Systemic (liver-tropic) | Moderate (lipid) | High | Scalable in vivo delivery |
| Virus-Like Particles (VLPs) | Pre-assembled RNP | 10-50% (various) | Systemic | Low (if PEGylated) | Moderate | Transient activity, reduced off-targets |
| Polymeric Nanoparticles | DNA, mRNA, or RNP | 15-45% (solid tumor cells) | Local/targeted | Variable | High | Tunable release, targeting |
Detailed Experimental Protocols for Key Delivery Applications
Protocol 1: ABE RNP Delivery to Primary Human T Cells via Electroporation
This protocol is optimized for high-efficiency, transient ABE delivery to minimize off-target effects and cellular toxicity.
Reagents & Materials: Primary human T cells, ABE protein (commercially purified or in-house), in vitro-transcribed sgRNA, P3 Primary Cell 96-well Nucleofector Kit (Lonza), Opti-MEM reduced serum media, IL-2 cytokine.
- Prepare RNP Complex: Resuspend ABE protein in sterile Cas9 buffer. Complex with sgRNA at a 1:1.2 molar ratio (e.g., 100 pmol ABE:120 pmol sgRNA). Incubate at 25°C for 10 minutes.
- Prepare Cells: Isolate and activate T cells using CD3/CD28 beads for 48-72 hours. On the day of nucleofection, count cells and resuspend in pre-warmed Opti-MEM at 1x10^7 cells/mL.
- Nucleofection: For each reaction, mix 20 µL cell suspension (2x10^5 cells) with 5 µL pre-complexed RNP in a nucleofection cuvette. Use the preset "EO-115" program on the 4D-Nucleofector system.
- Recovery: Immediately add 80 µL of pre-warmed complete media (with IL-2) to the cuvette. Transfer cells to a 96-well plate. Add an additional 150 µL of media. Incubate at 37°C, 5% CO2.
- Analysis: Assess viability at 24h (trypan blue). Harvest cells at 72-96h for genomic DNA extraction and NGS analysis of the target locus.
Protocol 2:In VivoABE Delivery to Mouse Liver via Lipid Nanoparticles (LNPs)
This protocol details LNP formulation for encapsulating ABE mRNA and sgRNA for hepatic editing.
Reagents & Materials: ABE mRNA (pseudouridine-modified), sgRNA (chemically modified), ionizable lipid (e.g., DLin-MC3-DMA), DSPC, cholesterol, PEG-lipid, ethanol, sodium acetate buffer (pH 4.0), dialysis cassettes, PBS.
- Formulation: Prepare an ethanol phase containing ionizable lipid, DSPC, cholesterol, and PEG-lipid at a defined molar ratio (e.g., 50:10:38.5:1.5). Prepare an aqueous phase of ABE mRNA and sgRNA in sodium acetate buffer (0.5 mg/mL total RNA).
- Mixing: Rapidly mix the ethanol and aqueous phases at a 1:3 volumetric ratio using a microfluidic mixer or turbulent pipetting. This induces spontaneous nanoparticle formation.
- Buffer Exchange: Dialyze the crude LNP suspension against PBS (pH 7.4) for 18 hours at 4°C to remove ethanol and establish a neutral pH.
- Characterization: Measure particle size (Zetasizer, target 70-100 nm), polydispersity index (PDI), and RNA encapsulation efficiency (RiboGreen assay).
- Administration & Analysis: Inject 200 µL of LNP formulation (1-3 mg/kg mRNA dose) intravenously into mice. Harvest liver tissue after 7-14 days. Isolate genomic DNA for NGS analysis of editing efficiency and indel background.
Visualization of Pathways and Workflows
ABE Delivery & Mechanism Workflow
Delivery Modality Decision Tree
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for ABE Delivery Optimization
| Reagent/Material | Primary Function in ABE Delivery | Key Considerations for Use |
|---|---|---|
| Nucleofector Kits (Lonza) | Enables electroporation of hard-to-transfect cells by combining cell-type specific buffers and electrical parameters. | Kit selection is cell-type critical (e.g., P3 for T cells, SG for HSCs). Optimize RNP concentration vs. viability. |
| Ionizable Cationic Lipids (e.g., DLin-MC3-DMA) | Core component of LNPs; enables mRNA encapsulation and efficient endosomal escape upon acidification in the endosome. | Formulation ratios with helper lipids are crucial for stability, efficiency, and tolerability. |
| AAV Serotypes (e.g., AAV9, AAV-DJ) | Viral capsids that confer tissue tropism for in vivo delivery of ABE DNA payloads. | Packing capacity is limited; often requires split-intein systems for full ABE. Pre-existing immunity is a concern. |
| Chemically Modified sgRNA | Enhances stability and reduces immunogenicity of sgRNA, especially for in vivo mRNA/LNP delivery. | 2'-O-methyl and phosphorothioate modifications at 3' and 5' ends are standard. |
| Purified ABE Protein | For RNP delivery, ensuring immediate activity and rapid degradation to minimize off-target windows. | Commercial sources vary in purity and formulation. In-house purification requires a His-tag or similar for FPLC. |
| Endosomal Escape Agents (e.g., Chloroquine) | Small molecule additives that buffer the endosome, promoting LNP or polymer payload release into the cytosol. | Can be cytotoxic. Used primarily for in vitro optimization to screen formulations. |
| PEGylated Lipids | Surface coating for LNPs/VLPs that reduces aggregation, improves circulation time, and modulates immunogenicity. | PEG length and density impact pharmacokinetics and potential for accelerated blood clearance. |
Adenine Base Editors (ABEs) are precise genome engineering tools that catalyze the direct, irreversible conversion of adenine (A) to guanine (G) in DNA without causing double-strand breaks. This capability enables the direct correction of pathogenic G•C to A•T mutations. The broader thesis on "How do adenine base editors work" necessitates a rigorous, quantitative assessment of their on-target editing efficiency, product purity, and unwanted byproducts. Next-Generation Sequencing (NGS) is the cornerstone for this quantification, making standardized benchmarking practices critical for comparing editors, optimizing conditions, and advancing therapeutic development.
Core Metrics for Quantifying Editing Outcomes
High-fidelity NGS analysis must move beyond simple "percent edited" calculations. The following key metrics, derived from processed sequencing data, provide a comprehensive profile of editor performance.
Table 1: Core Quantitative Metrics for ABE NGS Analysis
| Metric | Formula/Description | Interpretation & Importance |
|---|---|---|
| Editing Efficiency (%) | (Number of edited reads / Total aligned reads) * 100 | Primary measure of activity at the target locus. |
| Product Purity / Yield of Intended Product (%) | (Reads with only the intended A•T to G•C change / Total edited reads) * 100 | Critical for therapeutic applications; assesses precision. |
| Indel Frequency (%) | (Reads with insertions or deletions / Total aligned reads) * 100 | Measures genotoxic byproducts from nicking or off-target activity. |
| Strand Bias | Ratio of editing on the nicked vs. non-nicked strand. | Reveals mechanistic insights into repair kinetics and editor processivity. |
| Transition/Transversion Profile | Percentage of each nucleotide substitution type among all edited reads. | ABEs should primarily produce A>G (T>C) transitions; other substitutions indicate deaminase-independent noise or sequencing errors. |
| Average Editing Window | The ~5-nt window within the protospacer where editing is most efficient (e.g., positions 4-9 for ABE8e). | Defines the effective operating space of the editor. |
Experimental Protocol: Targeted Amplicon-Sequencing for ABE Evaluation
This standard workflow is essential for generating the data to calculate the metrics in Table 1.
- Sample Generation: Transfect or transduce cells with ABE and sgRNA expression constructs. Include negative controls (no editor, no sgRNA).
- Genomic DNA Extraction: Harvest cells 72-96 hours post-delivery. Use a column- or bead-based kit for high-purity gDNA.
- PCR Amplification of Target Locus:
- Primer Design: Design primers with overhangs containing Illumina adapter sequences, typically 250-350 bp from the edited site.
- First-PCR: Use a high-fidelity polymerase (e.g., Q5, KAPA HiFi) in a minimal cycle program (≤25 cycles) to prevent chimera formation.
- Clean-up: Purify amplicons using SPRI beads.
- Indexing PCR (Second-PCR):
- Add dual-index barcodes (i5 and i7) and full sequencing adapters via a second, limited-cycle PCR.
- Pool barcoded samples equimolarly.
- Sequencing: Run on an Illumina MiSeq or NextSeq platform. Aim for >10,000x read depth per sample for robust quantification of low-frequency events (<0.1%).
- Data Analysis Pipeline:
- Demultiplexing: Assign reads to samples using barcode sequences.
- Alignment: Map reads to the reference genome (e.g., using BWA-MEM).
- Variant Calling: Use specialized, quantitative tools (see Table 2) to identify and quantify all nucleotide changes and indels at the target site.
Table 2: Essential NGS Analysis Software Tools
| Tool Name | Primary Function | Key Feature for Base Editing |
|---|---|---|
| CRISPResso2 | Quantifies editing from NGS amplicons. | Built-in base editing mode; decomposes outcomes into precise substitution combinations. |
| Breseq | Analyzes microbial NGS data for variants. | Excellent for plasmid-based editing studies in bacteria. |
| DeepEditor | Deep learning-based analysis tool. | Accurately quantifies complex, heterogeneous editing outcomes. |
| Geneious | Commercial GUI-based bioinformatics suite. | User-friendly visualization and variant calling for single targets. |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Key Reagents for ABE NGS Workflow
| Item | Function & Importance |
|---|---|
| High-Fidelity DNA Polymerase (e.g., NEB Q5, KAPA HiFi) | Minimizes PCR errors during amplicon generation, ensuring sequencing variants reflect true editing. |
| SPRI Beads (e.g., AMPure XP) | For size-selective cleanup of PCR amplicons, removing primers, dimers, and non-specific products. |
| Dual-Indexed Sequencing Adapters (e.g., Illumina Nextera XT) | Enables multiplexing of hundreds of samples in a single sequencing run, reducing cost per sample. |
| Validated ABE Expression Plasmid (e.g., ABE8e) | Standardized editor delivery vehicle for reproducible benchmarking across labs. |
| Positive Control gDNA Sample | Genomic DNA from a clonal line with a known, precisely edited allele. Essential for validating the entire NGS workflow's accuracy. |
| Negative Control gDNA Sample | gDNA from a non-edited but otherwise identically treated sample. Defines the baseline sequencing error rate and background noise. |
Visualization of Workflows and Relationships
Diagram 1: End-to-End NGS Workflow for ABE
Diagram 2: Core Metrics Derived from NGS Data
Advanced Considerations and Best Practices
- Error Correction: Employ unique molecular identifiers (UMIs) to tag original DNA molecules, allowing bioinformatic correction of PCR and sequencing errors.
- Multiplexing: Use pooled sgRNA libraries to benchmark multiple targets simultaneously, but validate key findings with individual, deep amplicon sequencing.
- Analysis Depth: For detecting very rare off-targets or indels (<0.1%), ultra-deep sequencing (>100,000x) is required, though amplicon-based methods may suffer from increased noise.
- Reporting: Always report the full analysis parameters, software versions, and minimum sequencing depth alongside quantitative results to enable replication and fair comparison across studies.
Robust benchmarking via standardized NGS analysis is fundamental to elucidating the mechanistic "how" of ABE function. By adhering to these best practices in experimental workflow, data analysis, and metric reporting, researchers can generate reliable, comparable data. This rigor accelerates the iterative optimization of ABE tools and builds a solid evidentiary foundation for their translation into therapeutics.
ABE Validation and Comparisons: How Do Current Editors Stack Up?
Within the broader research thesis on How do adenine base editors (ABEs) work, it is imperative to contextualize their mechanism, performance, and utility against their primary counterpart: cytosine base editors (CBEs). This comparative analysis provides an in-depth technical guide to the efficiency, precision, and application scope of these two foundational genome-editing tools, synthesizing current data and methodologies for research and therapeutic development.
Core Mechanisms and Architectures
Both ABEs and CBEs are fusion proteins comprising a catalytically impaired Cas9 (dCas9 or nickase Cas9) tethered to a nucleobase deaminase enzyme. Their fundamental divergence lies in the deaminase and its target.
- ABE (Adenine Base Editor): Evolved from E. coli TadA, ABEs catalyze the deamination of adenine (A) to inosine (I) in DNA, which is replicated as guanine (G), effecting an A•T to G•C conversion.
- CBE (Cytosine Base Editor): Derived from rat APOBEC1, CBEs catalyze the deamination of cytosine (C) to uracil (U), leading to a C•G to T•A conversion after replication.
Crucially, both systems incorporate an uracil DNA glycosylase inhibitor (UGI) in CBEs or tailored architectures in ABEs to prevent unwanted base-excision repair that would reduce editing efficiency or produce indels.
Diagram Title: Core Catalytic Mechanisms of ABE and CBE
Quantitative Efficiency and Precision Comparison
Efficiency (editing rate) and precision (product purity, indel rate) are critical metrics. The following table summarizes recent comparative data from studies in human cell lines.
Table 1: Comparative Performance of ABE and CBE
| Metric | ABE (e.g., ABE8e) | CBE (e.g., BE4max) | Notes & Context |
|---|---|---|---|
| Peak Editing Efficiency | 50-80% | 60-90% | Highly dependent on sequence context, delivery, and cell type. CBEs often show higher raw efficiency. |
| Typical On-Target Product Purity | High (>90% of edits are desired A-to-G) | Moderate to Low (often 60-80% C-to-T) | CBE outcomes include undesired edits (e.g., C-to-G, C-to-A) due to uracil processing. |
| Average Indel Formation | Very Low (<0.5%) | Low (~1-5%) | ABEs generally produce fewer double-strand breaks and indels. |
| Effective Editing Window | Positions 4-8 (protospacer, A in AGA motif) | Positions 3-10 (protospacer, C in NTC context) | Defined from the PAM-distal end. Window size and preference vary by editor variant. |
| Off-Target DNA Editing | Very Low | Moderate to High (RNA off-targets) | CBE's APOBEC1 can deaminate cytosines in single-stranded cellular RNA. ABE8e shows some RNA off-target activity. |
| Sequence Context Bias | Strong preference for A in AGA/TCA motifs | Preference for C in T/C-rich contexts (e.g., TCN) | Limits targeting scope. New variants aim to reduce this bias. |
Scope and Application Landscape
The choice between ABE and CBE is dictated by the desired nucleotide conversion and the sequence context of the target site.
Table 2: Genomic Correction Scope & Therapeutic Applications
| Conversion Type | Primary Editor | Pathogenic Mutation Examples | Therapeutic Scope |
|---|---|---|---|
| A•T → G•C | ABE | TMC1 c.1253A>G (Hearing loss), FANCONI ANEMIA mutations, SERPINA1 (PiZ) | Corrects ~47% of known pathogenic point mutations requiring a transversion. |
| C•G → T•A | CBE | TP53 driver mutations, HEXA (Tay-Sachs), PRNP (prion disease), APOE4 (Alzheimer's risk) | Corrects ~14% of pathogenic point mutations (primarily C•G to T•A transitions). |
| C•G → G•C | Dual-acting or CBE-derived | Some sickle-cell disease variants | Emerging editors (e.g., glycosylase-base editors) can produce transversions. |
Detailed Experimental Protocol for Side-by-Side Evaluation
The following protocol outlines a standardized method to directly compare ABE and CBE efficiency and outcomes at matched genomic loci.
Objective: Quantify and compare editing efficiency, product distribution, and indel rates for ABE and CBE at multiple target sites.
Materials & Workflow:
Diagram Title: Workflow for Comparative ABE/CBE Evaluation
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents for ABE/CBE Research
| Reagent / Material | Function & Description | Example Vendor/ID (for reference) |
|---|---|---|
| ABE Expression Plasmid | Encodes the adenine deaminase-dCas9(nCas9) fusion protein. Critical for A-to-G editing. | Addgene #138489 (ABE8e) |
| CBE Expression Plasmid | Encodes the cytosine deaminase-dCas9(nCas9)-UGI fusion protein. Critical for C-to-T editing. | Addgene #136951 (BE4max) |
| sgRNA Cloning Vector | Backbone for expressing single guide RNA under a U6 promoter. | Addgene #135436 (pGL3-U6-sgRNA) |
| High-Efficiency Transfection Reagent | For delivering plasmid DNA to mammalian cells (e.g., HEK293T, iPSCs). | PEI MAX, Lipofectamine 3000 |
| NGS Amplicon-Seq Kit | For preparing deep sequencing libraries from purified PCR amplicons. | Illumina DNA Prep, Nextera XT |
| Genomic DNA Extraction Kit | For clean gDNA isolation from transfected cells for PCR analysis. | Qiagen DNeasy Blood & Tissue Kit |
| CRISPR Analysis Software | Computational tool to quantify base editing and indels from NGS data. | CRISPResso2, BE-Analyzer |
ABEs and CBEs are complementary technologies with distinct profiles. ABEs offer superior product purity and lower indel rates, making them attractive for therapeutic applications where precision is paramount. CBEs offer higher raw efficiency and a broader targeting window but require careful monitoring of sequence context and off-target effects. The ongoing research thesis on ABE mechanism continues to drive the engineering of next-generation editors with expanded targeting scope (e.g., dual A/C editors), reduced sequence context bias, and minimized off-target activity, pushing the entire field toward more versatile and safer genome medicine.
Advancements in gene editing have transitioned from creating double-strand breaks to precise, single-base changes. Adenine Base Editors (ABEs) exemplify this shift, enabling the direct, irreversible conversion of an A•T base pair to a G•C pair without requiring double-stranded DNA breaks or donor DNA templates. This whitepaper evaluates the next-generation ABE8 series—specifically ABE8e, ABE8s, and ABE8.20 variants—within the broader thesis on How do adenine base editors (ABEs) work? The evolution from ABE7.10 to the ABE8 series represents a significant leap in efficiency and application potential, driven by extensive protein engineering. This guide provides a technical deep dive into their mechanisms, performance data, and experimental protocols for researchers and therapeutic developers.
Core Mechanism and Evolution of ABE8 Variants
All ABEs are fusion proteins comprising a catalytically impaired CRISPR-Cas nickase (most commonly nSpCas9) tethered to an engineered adenine deaminase enzyme (TadA). The TadA domain catalyzes the deamination of adenine (A) to inosine (I) in single-stranded DNA within the R-loop formed by Cas9 binding. Inosine is read as guanine (G) by cellular polymerases, resulting in an A•T to G•C conversion during DNA replication or repair. The nickase activity of Cas9 nicks the non-edited strand, biasing cellular repair to use the edited strand as a template, thereby increasing editing efficiency.
The ABE8 series was developed through extensive directed evolution of the TadA domain to enhance activity and substrate scope. Key variants include:
- ABE8e: Incorporates eight mutations (L84F, D108N, A106V, D147Y, E155V, Q154R, S146C, K160S) in TadA-8e. It exhibits dramatically higher on-target editing efficiency but may have increased off-target effects.
- ABE8s: Incorporates seven mutations (L84F, D108N, A106T, D147Y, E155V, K160S, R152P) in TadA-8s. It is engineered for a tighter activity window and reduced off-target RNA editing compared to ABE8e, offering a better efficiency/specificity balance.
- ABE8.20: A more recent variant featuring TadA-8.20, evolved for further reduced off-target DNA and RNA editing while maintaining robust on-target activity, making it a leading candidate for therapeutic applications.
Quantitative Performance Comparison
The following tables summarize key performance metrics for ABE8 variants compared to the previous benchmark, ABE7.10.
Table 1: On-Target Editing Efficiency & Product Purity
| Editor Variant | Avg. On-Target Efficiency* (%) | Transition Purity (A•T to G•C) (%) | Typical Activity Window (Position within Protospacer) | Primary TadA Domain |
|---|---|---|---|---|
| ABE7.10 | 10-50 | >99.9% | Positions 4-8 (most efficient at A5-A7) | TadA-7.10 |
| ABE8e | 40-95 | >99.9% | Positions 4-10 (broadened) | TadA-8e |
| ABE8s | 30-80 | >99.9% | Positions 4-9 | TadA-8s |
| ABE8.20 | 35-85 | >99.9% | Positions 4-8 | TadA-8.20 |
*Efficiency varies significantly by target locus and cell type. Ranges are compiled from multiple studies.
Table 2: Specificity and Other Key Metrics
| Editor Variant | Off-Target DNA Editing* | Off-Target RNA Editing* | Processivity (Multiple edits per binding event) | Catalytic Rate (kcat) Improvement vs. ABE7.10 |
|---|---|---|---|---|
| ABE7.10 | Low | Low to Moderate | Low | 1x (Baseline) |
| ABE8e | High | Very High | High | ~1100x |
| ABE8s | Moderate | Low | Moderate | ~590x |
| ABE8.20 | Very Low | Undetectable | Moderate | ~590x (with enhanced fidelity) |
*Relative assessment based on current literature (e.g., CIRCLE-seq, RNA-seq data).
Experimental Protocols for Evaluation
Protocol 1: Measuring On-Target Editing Efficiency via Next-Generation Sequencing (NGS)
Objective: Quantify A•T to G•C conversion frequency at a specific genomic locus. Methodology:
- Delivery: Transfect HEK293T cells (or target cell line) with plasmids encoding the ABE variant (e.g., ABE8.20) and a specific sgRNA using a preferred method (e.g., PEI, lipofection, nucleofection).
- Harvest: At 72 hours post-transfection, harvest cells and extract genomic DNA using a commercial kit.
- PCR Amplification: Design primers flanking the target site (amplicon size: 250-500 bp). Perform PCR to amplify the locus from ~100 ng of genomic DNA.
- Library Prep & Sequencing: Purify PCR products, add Illumina sequencing adapters via a second limited-cycle PCR, and purify the final library. Quantify and pool for NGS on a platform like MiSeq.
- Analysis: Use bioinformatics tools (e.g., CRISPResso2, BE-Analyzer) to align reads and calculate the percentage of sequences containing the intended A-to-G conversion.
Protocol 2: Assessing Off-Target DNA Editing via CIRCLE-seq
Objective: Genome-wide identification of potential off-target sites. Methodology:
- Genomic DNA Isolation & Digestion: Isolate high-molecular-weight genomic DNA from untreated cells. Digest with a restriction enzyme that leaves 5' overhangs.
- Circularization: Dilute and ligate digested DNA with T4 DNA ligase to form circular DNA molecules.
- Cas9 Cleavage In Vitro: Incubate circularized DNA with the corresponding nSpCas9 protein complexed with the sgRNA of interest. This linearizes circles only at sites complementary to the sgRNA.
- Library Preparation: Repair ends of linearized DNA, add adapters, and amplify by PCR for NGS.
- Analysis: Map sequencing reads to the reference genome. Sites of enrichment correspond to Cas9 binding/cleavage sites, indicating potential off-target loci for the ABE. These sites should be validated by targeted amplicon sequencing.
Visualizing ABE Mechanism and Workflow
Diagram 1: ABE8 Mechanism from Binding to Edit
Diagram 2: ABE8 Evaluation Experimental Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
| Reagent / Material | Function & Explanation |
|---|---|
| ABE8 Expression Plasmids | Plasmids (e.g., pCMV-ABE8.20) encoding the base editor and a U6-driven sgRNA scaffold for mammalian delivery. Essential for transient expression in cells. |
| Chemically Modified sgRNAs | Synthetic sgRNAs with 2'-O-methyl-3'-phosphorothioate modifications at terminal nucleotides. Enhance stability, RNP formation, and editing efficiency, especially in primary cells. |
| RNP Complexes | Pre-assembled complexes of purified ABE8 protein and sgRNA. Enable rapid editing with reduced off-target effects and shorter exposure times compared to plasmid delivery. |
| HEK293T Cell Line | A robust, easily transfected adherent cell line. The standard workhorse for initial benchmarking and efficiency comparisons of novel ABE variants. |
| Nucleofection/Kits | Electroporation-based transfection systems (e.g., Lonza Nucleofector) and optimized kits for delivering RNPs or plasmids into hard-to-transfect primary and stem cells. |
| High-Fidelity DNA Polymerase | Enzyme (e.g., Q5, KAPA HiFi) for accurate amplification of target loci from genomic DNA prior to NGS, minimizing PCR-introduced errors. |
| NGS Library Prep Kit | Kits specifically designed for amplicon sequencing (e.g., Illumina DNA Prep) to prepare barcoded libraries from PCR amplicons for high-throughput sequencing. |
| CIRCLE-seq Kit | Commercial or published protocol reagents for performing genome-wide, unbiased off-target DNA editing profiling. |
| CRISPResso2 / BE-Analyzer | Bioinformatics software packages specifically designed to quantify base editing outcomes from NGS data, reporting efficiency, product purity, and indel rates. |
Within the broader thesis on understanding how adenine base editors (ABEs) work, a critical component is assessing their precision. ABEs, which catalyze the direct conversion of A•T to G•C base pairs without generating double-stranded DNA breaks, hold immense therapeutic potential. However, off-target editing, whether at the DNA or RNA level, poses significant safety risks. Therefore, rigorous, genome-wide specificity benchmarking is a non-negotiable step in their characterization and development. This guide details two pivotal, high-sensitivity methods: GOTI and Digenome-seq.
Genome-Wide Off-Target Analysis by Two-Cell Embryo Injection (GOTI)
GOTI is an in vivo method designed to detect off-target edits with single-nucleotide resolution and minimal background noise. Its core innovation is the use of mouse embryonic cells from the same embryo as isogenic experimental and control samples.
Experimental Protocol for GOTI with ABEs
- Embryo Generation: Generate embryos from a mouse line with a ubiquitous fluorescent marker (e.g., ACTB-tdTomato).
- Two-Cell Stage Injection: At the two-cell stage, inject one blastomere with two components:
- ABE Expression Construct: Plasmid or mRNA encoding the adenine base editor (e.g., ABE8e).
- Cre Recombinase mRNA: To activate tdTomato expression in the injected cell and its progeny.
- Embryo Culture: Culture embryos to the post-implantation epiblast stage (~E9.5).
- Cell Sorting: Dissociate the embryonic cells and use Fluorescence-Activated Cell Sorting (FACS) to separate tdTomato-positive (edited) and tdTomato-negative (internal control) cell populations from the same embryo.
- Whole-Genome Sequencing (WGS): Perform deep whole-genome sequencing (>50X coverage) on the genomic DNA from both sorted populations.
- Bioinformatic Analysis: Use specialized variant callers (e.g., GATK) to identify A-to-G (or T-to-C on the opposite strand) single-nucleotide variants (SNVs). Filter out SNVs present in both the experimental and control samples, as these are likely germline polymorphisms or sequencing errors. Off-target sites are defined as A-to-G SNVs unique to the tdTomato-positive (edited) sample.
Workflow for GOTI Specificity Profiling
Digenome-seq (Digested Genome Sequencing)
Digenome-seq is a highly sensitive in vitro method that identifies off-target sites by detecting nuclease-induced breaks in genomic DNA. While originally for nucleases, it has been adapted for base editors by leveraging the nickase activity of Cas9n (D10A) used in many ABE architectures or by incorporating a repair enzyme that creates breaks at edited sites.
Experimental Protocol for Digenome-seq with ABEs
- Genomic DNA Extraction: Isolate high-molecular-weight genomic DNA from target cells (e.g., human cell line).
- In Vitro Editing: Incubate the purified genomic DNA with the ABE ribonucleoprotein (RNP) complex (purified ABE protein + sgRNA) under optimal reaction conditions.
- Whole-Genome Sequencing: Shear the edited and control (untreated) genomic DNA and prepare WGS libraries. Sequence at very high depth (>100X).
- Bioinformatic Analysis: Map sequencing reads to the reference genome. For standard nickase-based ABEs, use tools like Digenome-seq 2.0 to identify sites with a localized increase in mismatches (A-to-G conversions) and a depletion of read coverage, indicating a nick or break. For modified protocols using repair enzymes (e.g., EndoV or EndoVIII for A-to-I mismatches), identify sites with abrupt truncations of sequencing reads.
Workflow for Digenome-seq Analysis
Comparative Analysis of Methods
| Feature | GOTI | Digenome-seq |
|---|---|---|
| System | In vivo (mouse embryo) | In vitro (purified genomic DNA) |
| Biological Context | High (native chromatin, repair systems) | Low (naked DNA, no cellular context) |
| Primary Detection Signal | A-to-G SNVs in sequenced DNA | Localized mismatches/coverage drops |
| Background Noise | Very low (isogenic control) | Low (untreated DNA control) |
| Throughput & Cost | Lower throughput, higher cost | Higher throughput, lower cost |
| Key Advantage | Reveals cell-type specific, in vivo off-targets | Unbiased, genome-wide, extremely sensitive |
| Limitation | Technically complex, not human genome | May overpredict sites not active in cells |
The Scientist's Toolkit: Research Reagent Solutions
| Reagent/Material | Function & Importance |
|---|---|
| ABE Expression Plasmid/mRNA | Delivery of the base editor. mRNA allows rapid, transient expression without genomic integration. |
| High-Fidelity Cas9 Variant (e.g., ABE8e-NGG) | The core editor. Using a high-fidelity Cas9 domain is crucial to minimize sgRNA-dependent off-targets. |
| Chemically Modified sgRNA | Enhances stability and can reduce off-target binding. Critical for in vitro Digenome-seq. |
| Ultra-Pure Genomic DNA Kit | Essential for Digenome-seq to obtain high-molecular-weight DNA without contaminants that inhibit the in vitro reaction. |
| Next-Generation Sequencing Kit (WGS) | For high-depth sequencing. Must provide uniform coverage and accurate base calling for SNV detection. |
| Variant Calling Pipeline (e.g., GATK, BCFtools) | Specialized bioinformatics software to distinguish true off-target edits from sequencing errors and polymorphisms. |
| Mouse Strain (e.g., ACTB-tdTomato) | For GOTI, provides the genetic basis for fluorescent labeling and sorting of edited cell lineages. |
Within the broader thesis on How do adenine base editors (ABEs) work, functional validation is the critical final step. It moves beyond confirming the presence of an A•T to G•C edit to demonstrating that the edit restores normal cellular protein function and phenotype. This whitepaper details core assays for validating phenotypic correction and protein function following ABE-mediated gene editing.
Core Validation Assays: From DNA to Phenotype
A robust validation pipeline assesses edits at multiple biological levels.
Table 1: Tiered Functional Validation Assay Suite
| Validation Tier | Assay Category | Specific Examples | Key Readout | Quantitative Metric |
|---|---|---|---|---|
| DNA & RNA | Sequencing | NGS (amplicon), Sanger | Edit efficiency, purity | % editing frequency, % indels |
| Protein | Expression & Localization | Western Blot, Immunofluorescence, Flow Cytometry | Protein presence, size, cellular location | Protein intensity, % positive cells |
| Protein | Biochemical Activity | Enzyme activity assay, Protein-binding assay (SPR, ELISA) | Catalytic function, ligand interaction | Vmax/Km, Binding affinity (KD) |
| Cellular Phenotype | Rescue/Correction | Cell viability, Morphology, Reporter assays | Restoration of normal function | IC50, Colony count, Fluorescence units |
| Organismal Phenotype | In Vivo Function | Animal model disease readout | Physiological correction | Survival rate, Behavioral score |
Detailed Experimental Protocols
Protocol 1: NGS-Based Editing Efficiency and Purity Analysis
Objective: Quantify A-to-G conversion efficiency and byproducts.
- Primer Design: Design primers to amplify a ~300-400bp region flanking the target site.
- PCR Amplification: Perform two rounds of PCR. Round 1: Amplify genomic DNA. Round 2: Attach Illumina sequencing adapters and sample barcodes.
- Library Purification & Quantification: Use bead-based purification. Quantify via qPCR.
- Sequencing: Run on an Illumina MiSeq or similar platform.
- Analysis: Align reads to reference genome. Use tools like CRISPResso2 or BE-Analyzer to calculate: % A-to-G conversion, % other base conversions, and % indel frequency.
Protocol 2: Protein Function Rescue via Cell Viability Assay
Objective: Validate correction of a pathogenic variant conferring drug sensitivity.
- Cell Line: Use isogenic disease model cells (e.g., harboring a pathogenic SNV).
- Editing: Transfect with ABE (e.g., ABE8e) + sgRNA or treat with RNP.
- Selection: Apply relevant drug (e.g., 6-thioguanine for HPRT1 correction) 72h post-editing.
- Culture: Maintain drug selection for 7-10 days.
- Analysis: Fix and stain colonies with crystal violet. Count colonies. Calculate correction frequency as: (Colonies from edited sample / Colonies from wild-type control) x 100%.
Protocol 3: Direct Protein Activity Assay (e.g., Enzymatic)
Objective: Measure restoration of enzymatic activity from a corrected allele.
- Lysate Preparation: Lyse edited cells 5-7 days post-editing in non-denaturing buffer.
- Protein Normalization: Determine total protein concentration (Bradford assay).
- Reaction Setup: Incubate normalized lysates with enzyme-specific substrate. Include negative (uncorrected) and positive (wild-type) controls.
- Kinetic Measurement: Monitor product formation spectrophotometrically or fluorometrically over time.
- Analysis: Calculate specific activity (product formed/min/µg protein). Report % activity restored relative to wild-type control.
Visualizing Validation Workflows
Functional Validation Cascade for ABE Editing
ABE Mechanism to Functional Correction
The Scientist's Toolkit: Key Research Reagents & Materials
Table 2: Essential Reagents for ABE Functional Validation
| Reagent/Material | Function | Example Product/Catalog |
|---|---|---|
| ABE Plasmid or Protein | Core editor delivery. | pCMV_ABE8e (Addgene #138495), HiFi S.p. ABE8e (NEB) |
| Validated sgRNA | Targets editor to genomic locus. | Chemically synthesized sgRNA, Custom gRNA expression plasmid. |
| Next-Generation Sequencing Kit | Quantifying edit efficiency and purity. | Illumina MiSeq Reagent Kit v3, Amplicon-EZ service (Genewiz). |
| Antibodies (Specific) | Detecting protein expression/localization. | Anti-[target protein] antibody for WB/IF; must distinguish wild-type vs. mutant if possible. |
| Activity Assay Kit | Measuring restored biochemical function. | β-galactosidase, Luciferase, or specific enzyme activity kits (e.g., Promega, Abcam). |
| Cell Viability/Phenotype Assay | Measuring cellular rescue. | CellTiter-Glo (viability), Crystal violet (clonogenic), High-content imaging reagents. |
| Positive Control gDNA/cDNA | Baseline for functional assays. | Genomic DNA from wild-type/isogenic control cell line. |
| Negative Control sgRNA | Controls for off-target effects. | Non-targeting scrambled sgRNA. |
Adenine Base Editors (ABEs) enable the direct, irreversible conversion of A•T to G•C base pairs without inducing double-strand DNA breaks, positioning them as transformative tools for precise gene correction. Within the broader thesis on "How do adenine base editors (ABEs) work?", this analysis focuses on the critical translational bridge: systematically evaluating current safety limitations and outlining the experimental and engineering strategies required to advance ABEs toward clinical application.
Current Safety Limitations: Quantitative Analysis
The clinical translation of ABEs is contingent upon overcoming key safety hurdles, primarily concerning off-target editing and unintended on-target byproducts. Recent studies provide the following quantitative data:
Table 1: Quantified Safety Limitations of Current ABE Systems
| Limitation Category | Specific Metric | Reported Incidence/Frequency | Detection Method | Primary Study (Year) | |
|---|---|---|---|---|---|
| DNA Off-Target Editing | Guide-independent off-targets (Cas9-dependent) | Up to 20 off-target sites with >0.1% frequency in certain cell types | CIRCLE-seq, Digenome-seq | (2023) | |
| Guide-dependent off-targets | Varies widely by guide; can exceed on-target efficiency in rare cases | CHANGE-seq, OFF-seq | (2024) | ||
| RNA Off-Target Editing | Transcriptome-wide adenosine deamination | Thousands of sites, though often at low efficiency (<0.1%) | RNA-seq, A-to-I RNA SNV calling | (2023) | |
| On-Target Byproducts | Stochastic A-to-I (inosine) formation | Can lead to A-to-C, A-to-T transversions at low frequencies (0.1-1%) | High-fidelity next-generation sequencing (NGS) | (2024) | |
| Bystander Editing | Editing of adjacent adenosines within the editing window (typically ~5 nt) | Frequency can reach >50% for some targets | Targeted NGS | (2023) | |
| Delivery-Associated Risks | Immunogenicity to bacterial TadA | Anti-TadA antibodies detected in ~70% of human sera samples | ELISA, immunoblot | (2022) | |
| Preexisting anti-Cas9 immunity | ~50-60% of individuals have circulating antibodies | Protein microarray | (2023) |
Experimental Protocols for Safety Assessment
A rigorous, multi-layered experimental framework is mandatory for characterizing ABE safety profiles.
Protocol 1: Comprehensive DNA Off-Target Analysis using CHANGE-seq
- Objective: Identify both guide-dependent and guide-independent off-target sites genome-wide.
- Materials: Purified ABE ribonucleoprotein (RNP) complex (e.g., ABE8e-nSpCas9), genomic DNA from target cells, CHANGE-seq adapter kit, NGS platform.
- Methodology:
- In Vitro Cleavage: Incubate 1 µg of genomic DNA with 200 nM ABE RNP in reaction buffer (37°C, 3 hours).
- Blunt-End Ligation: Purify DNA and ligate double-stranded adapters to the exposed ends generated by in vitro nicking/cleavage events.
- Library Amplification: Perform PCR amplification to create sequencing libraries.
- Bioinformatic Analysis: Sequence and map reads to the reference genome. Identify sites of adapter integration, which indicate Cas9-induced DNA strand discontinuities. Calculate off-target score for each site.
Protocol 2: Quantifying On-Target Bystander Editing and Byproducts
- Objective: Precisely measure the spectrum and frequency of all editing outcomes at the intended target locus.
- Materials: Genomic DNA from edited cells, locus-specific PCR primers, barcoded NGS library prep kit.
- Methodology:
- Amplicon Sequencing: Design primers to amplify a ~300 bp region flanking the target site. Perform PCR with unique molecular identifiers (UMIs).
- Deep Sequencing: Sequence to high coverage (>100,000x) using a 2x300 bp MiSeq run.
- Analysis Pipeline: Use tools like CRISPResso2 or BE-Analyzer to align reads and quantify the percentage of reads containing A-to-G conversions at the target adenosine, A-to-X changes at adjacent bystander adenosines, and other nucleotide substitutions (A-to-C/T).
Strategic Pathways to Enhanced Safety
The future clinical outlook relies on parallel engineering strategies to mitigate the risks quantified above.
Diagram Title: Safety Limitation Mitigation Pathways for Clinical ABEs
The Scientist's Toolkit: Key Reagent Solutions
Table 2: Essential Research Reagents for ABE Safety Profiling
| Reagent / Material | Provider Examples | Primary Function in Safety Research |
|---|---|---|
| High-Fidelity ABE Plasmids | Addgene (ABE8e, ABE9, ABE7.10f), custom synthesis | Source of editor cDNA for generating improved variants with reduced off-target activity. |
| Purified ABE RNP Complex | Integrated DNA Technologies (IDT), Thermo Fisher Scientific, in-house purification | Ready-to-use complex for highly controlled in vitro and ex vivo experiments; reduces off-targets vs. plasmid delivery. |
| CIRCLE-seq Kit | Custom protocol; core NGS service providers | Comprehensive, amplification-free method for identifying Cas9-dependent DNA off-target sites genome-wide. |
| UMI-based Amplicon-Seq Kit | Illumina (TruSeq), Swift Biosciences | Enables accurate, error-corrected quantification of on-target editing outcomes and byproducts. |
| Humanized TadA Domain Constructs | Custom gene synthesis from Twist Bioscience, Genscript | Reduce immunogenicity by replacing bacterial TadA sequences with human-derived deaminase motifs. |
| Primary Human Immune Cells | STEMCELL Technologies, AllCells | For ex vivo assessment of ABE component immunogenicity and T-cell activation risks. |
| In Vivo Delivery Vehicles (LNP, AAV) | Precision NanoSystems (LNP), Vigene Biosciences (AAV) | Critical for assessing safety and efficacy of ABE delivery in preclinical animal models. |
The trajectory for ABE clinical translation is defined by a multi-parameter optimization problem: balancing efficiency, specificity, and tolerability. The future outlook necessitates:
- Combinatorial Engineering: Integrating high-fidelity Cas domains with evolved, RNA-specific TadA variants and humanized protein sequences into a single construct.
- Advanced Delivery: Developing transient, efficient, and tissue-specific delivery systems (e.g., improved LNPs) that limit exposure and immune detection.
- Rigorous In Vivo Toxicology: Establishing standardized, long-term studies in relevant animal models to rule out oncogenic risk from rare off-target events.
- Patient Screening: Implementing pre-treatment screening for preexisting immunity to guide patient stratification and mitigate adverse events.
The resolution of current limitations is not a barrier but an active design process. Through iterative engineering guided by the stringent experimental protocols outlined, ABEs are poised to evolve from powerful research tools into safe, effective, and broadly applicable genomic medicines.
Conclusion
Adenine base editors represent a transformative leap beyond conventional CRISPR-Cas9, enabling precise, efficient, and irreversible A•T to G•C conversion without requiring double-strand breaks. This article has detailed the sophisticated molecular design of ABEs, practical methodologies for their application across research and therapeutic contexts, strategies to overcome key experimental hurdles, and frameworks for rigorous validation. For the research and drug development community, mastering ABE technology is crucial for exploring genetic function and developing next-generation therapies for monogenic disorders caused by G•C to A•T mutations. Future directions hinge on developing editors with expanded targeting ranges (e.g., near-PAM-less variants), enhanced specificity profiles, and efficient in vivo delivery systems. As the field progresses, ongoing innovation in ABE engineering and rigorous pre-clinical validation will be paramount for translating this powerful genome-editing tool from the bench to the clinic, opening new avenues for curing genetic diseases.